Thioacetals and Corey-Seebach Intermediates
In this tutorial I want to talk about thioacetals, which are a really awesome functional group in organic chemistry.
They can do two amazing things. First, they can temporarily convert aldehydes and ketones from electrophiles into nucleophiles (known as the “umpolung strategy”), which opens a huge door for many chemical transformations. Second, they allow us to completely remove a carbonyl group under relatively mild conditions.
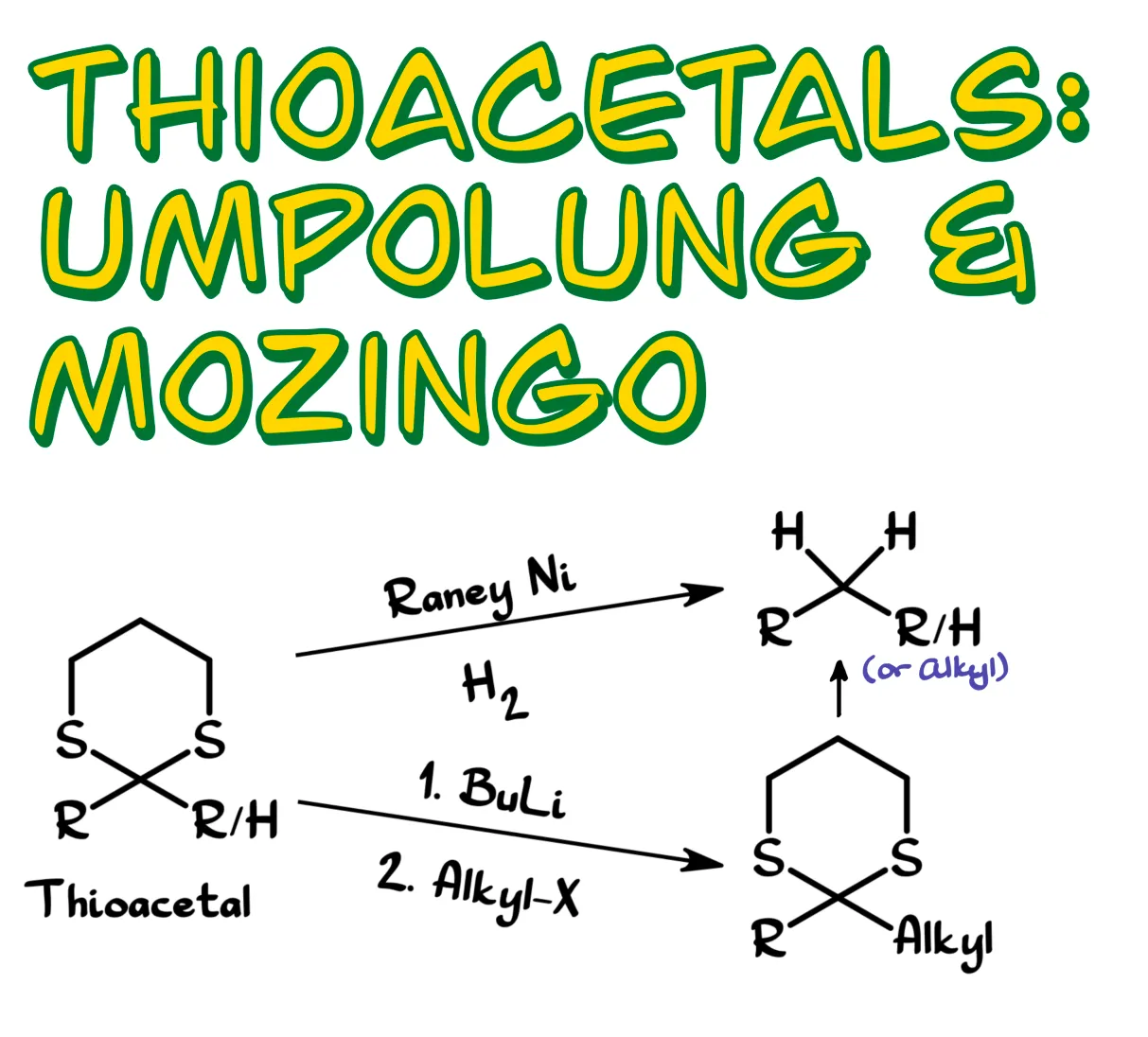
In this tutorial we will go through three main ideas. First, how thioacetals are formed. Then we will look at the reactions that thioacetals can undergo. Finally, we will discuss how to remove them once we no longer need them.
Thioacetal Formation
Similar to regular acetal formation, we begin with a carbonyl compound.
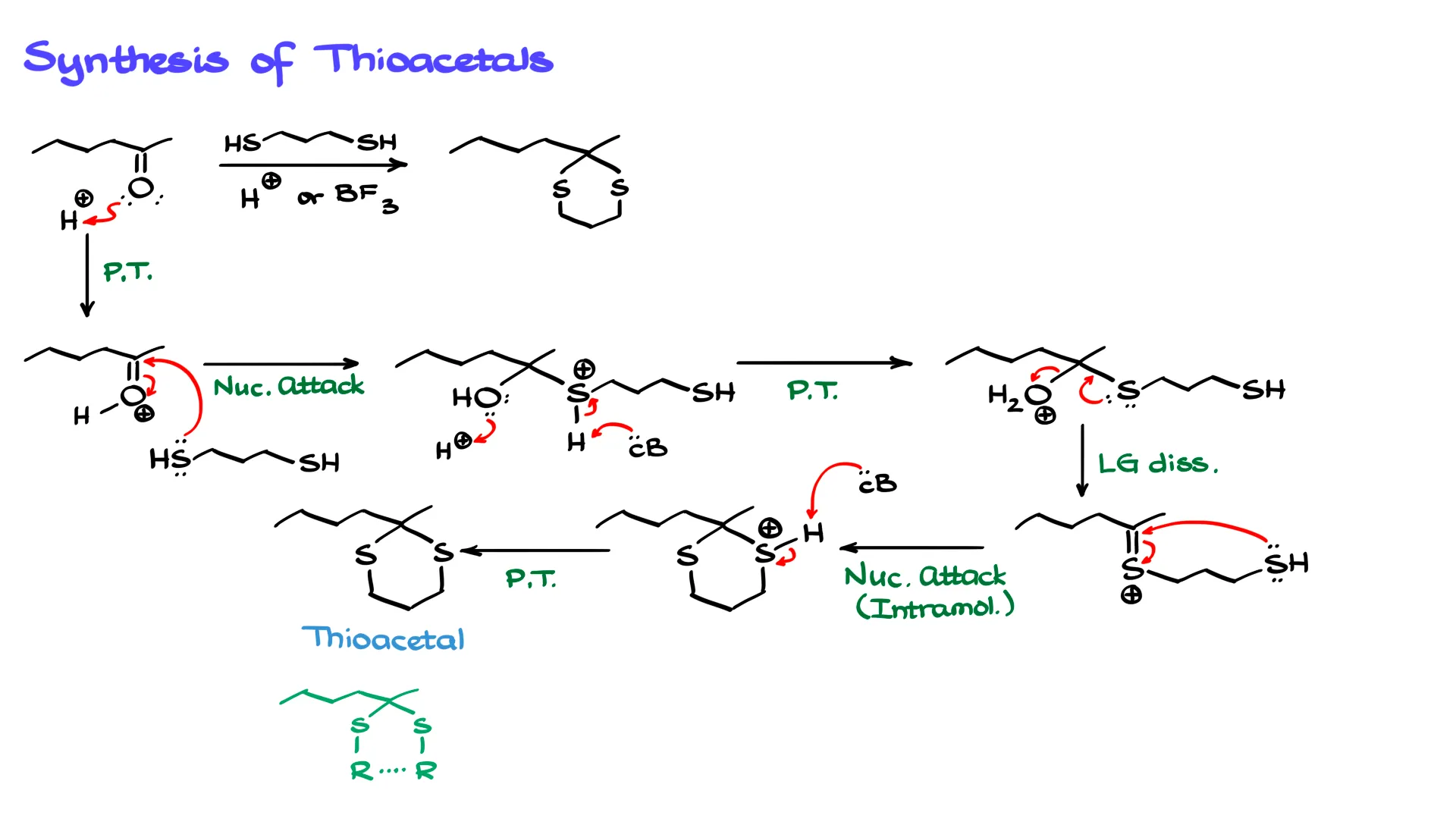
In this case I have a ketone, and we react it with a thiol or a dithiol like the one shown here. The reaction is catalyzed by either a Brønsted acid or a Lewis acid, with Lewis acids being somewhat more common. For simplicity, I will just use a regular acid in the mechanism.
The first step is a simple proton transfer. I bring in H⁺ and protonate the carbonyl oxygen. This gives us a protonated, or activated, carbonyl group.
From this point we perform a nucleophilic attack from the thiol. One of the sulfur atoms acts as a nucleophile and attacks the carbonyl carbon, forming a new carbon–sulfur bond and giving the corresponding intermediate.
Next we perform a couple of proton transfers. We need to remove the proton from the sulfur atom and protonate the OH group so that it becomes a better leaving group. Since this mechanism should already be familiar, I will combine these steps. A conjugate base removes the proton from sulfur, while another molecule of acid protonates the hydroxyl group.
After these proton transfers we obtain the next intermediate. Now the OH group has become a good leaving group, so sulfur can assist in its departure. The electron pair on sulfur pushes in, the leaving group departs as water, and we form a carbon–sulfur double bond intermediate.
This C=S bond is somewhat similar to a carbonyl in that it is electrophilic, and the carbon is positively polarized. That means the second sulfur atom in the molecule can act as a nucleophile. It attacks the electrophilic carbon, forming a cyclic intermediate.
At this point the only remaining step is to remove the proton. A conjugate base removes that proton and we obtain the final product, the thioacetal.
In this example the product is cyclic because we started with a dithiol. The same thing happens in regular acetal formation when we use a diol such as ethylene glycol.
However, thioacetals do not have to be cyclic. In a general structure the two sulfur atoms can each be attached to R groups, and those R groups may or may not be connected to each other.
So now we have our thioacetal. The next topic is one of the coolest things these compounds can do.
Corey-Seebach Intermediate (Reagent)
The hydrogen located on the carbon between the two sulfur atoms is somewhat acidic.
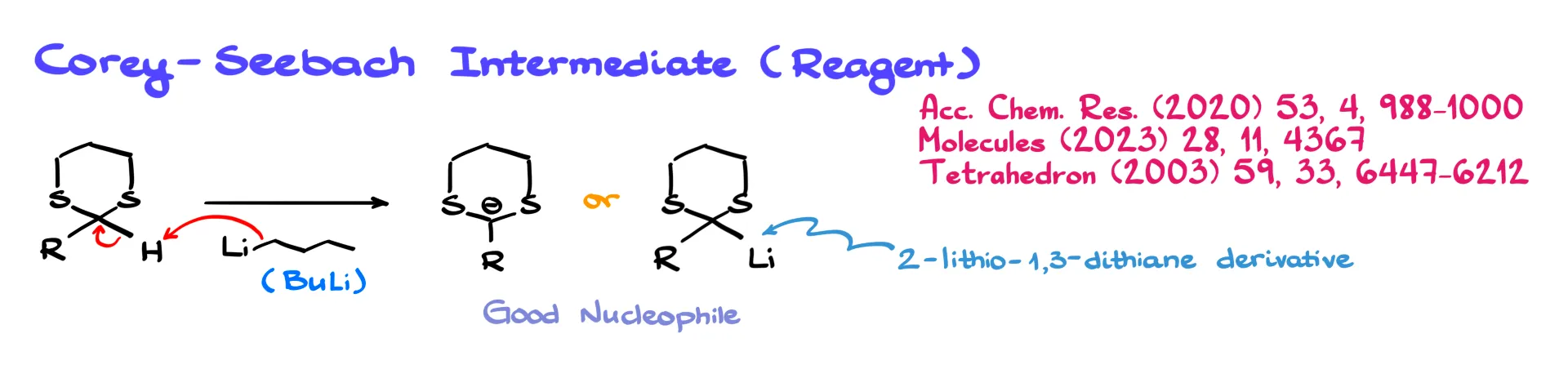
If we treat the molecule with a strong base such as butyllithium, the base can remove that proton. The product is best represented as a lithium–carbon bond rather than just a negatively charged carbon.
In the cyclic case, this intermediate is known as the Corey–Seebach reagent, named after Elias Corey and Dieter Seebach, who popularized this chemistry.
What makes this intermediate so powerful is that it is an excellent nucleophile. As a nucleophile it can react with many different electrophiles. In general, the lithium derivative reacts with an electrophile to form a new carbon–carbon bond between the thioacetal carbon and the electrophilic carbon.
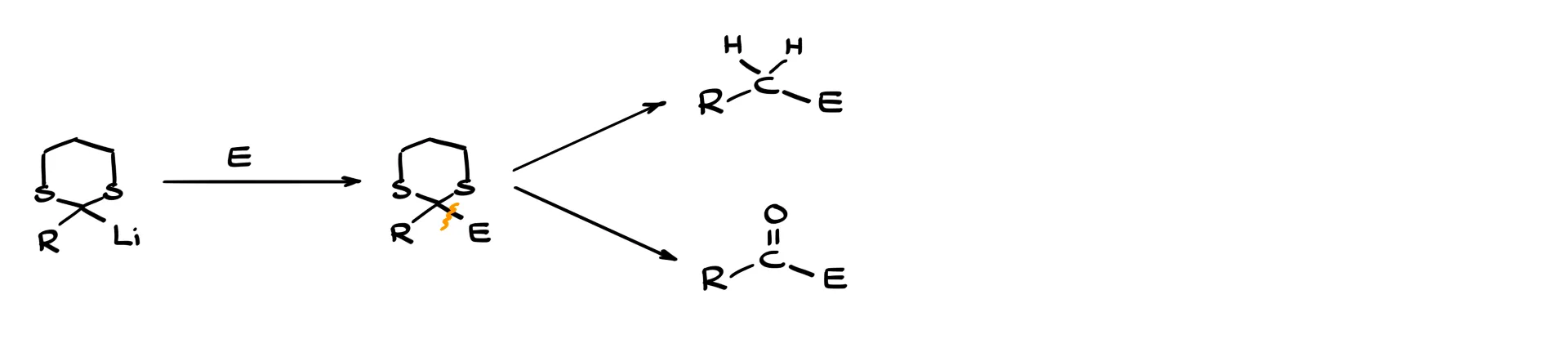
After that transformation we have two major options. We can either remove the sulfur completely, reducing the carbon to a CH₂ group, or we can hydrolyze the thioacetal and regenerate the carbonyl group.
Now let’s look at several types of electrophiles.
Reactions with Alkyl Halides and Similar Reagents
The first example is a simple alkyl halide, or related leaving-group derivatives such as sulfonate esters like triflates, mesylates, or tosylates.
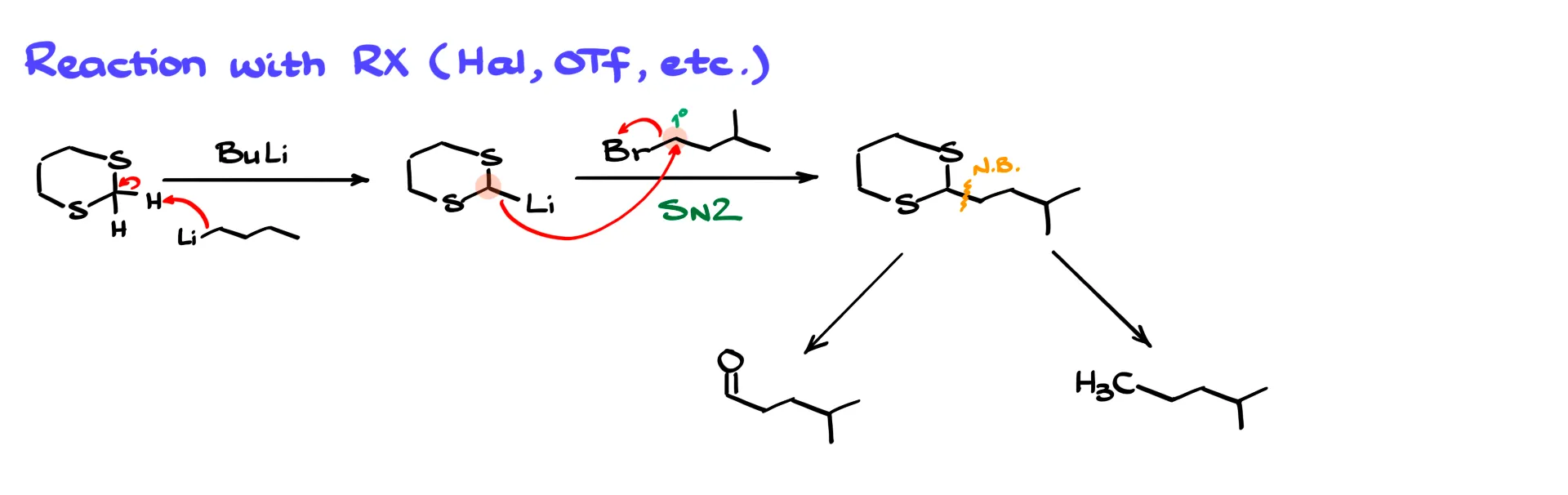
Starting with a thioacetal, we treat it with butyllithium to form the lithium derivative. Then we react it with an alkyl bromide. The nucleophile performs an SN2 reaction, displacing the leaving group and forming a new carbon–carbon bond.
From this point we can either remove the sulfur completely or hydrolyze the thioacetal. If we removed the sulfur in this specific example we would simply obtain a methyl group, which is not the most efficient way to install one, but it illustrates the concept. Alternatively, hydrolysis would give the corresponding aldehyde, which could be used in further carbonyl chemistry.
Let’s look at a literature example. We start with a thioacetal and treat it with butyllithium to form the lithium derivative. In the next step we react it with an electrophile that contains two primary chlorides: one allylic and one regular.
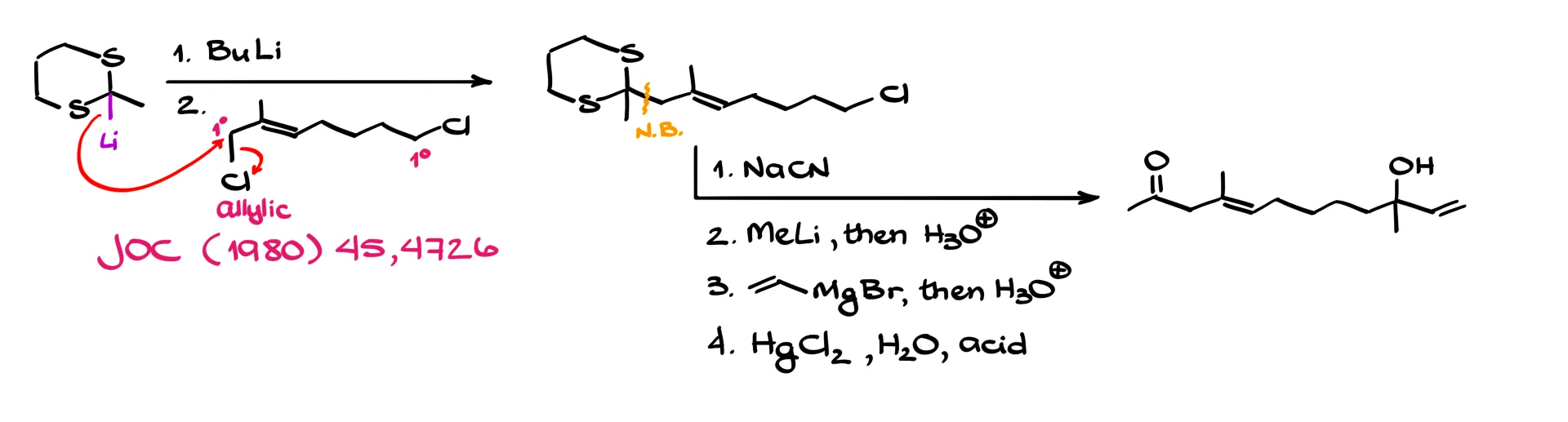
Because allylic positions are more reactive in substitution reactions, the nucleophile attacks the allylic carbon and displaces chloride. This forms a new carbon–carbon bond at that position.
From there, a sequence involving sodium cyanide, methyllithium followed by a Grignard reaction, and finally hydrolysis using a mercury salt leads to the final product. Most of the intermediates in this sequence should be familiar from sophomore organic chemistry, so it is a good exercise to work through the mechanisms and verify the final structure.
Reactions with Epoxides
Next, let’s talk about epoxides. Love them or hate them, epoxides are incredibly useful.
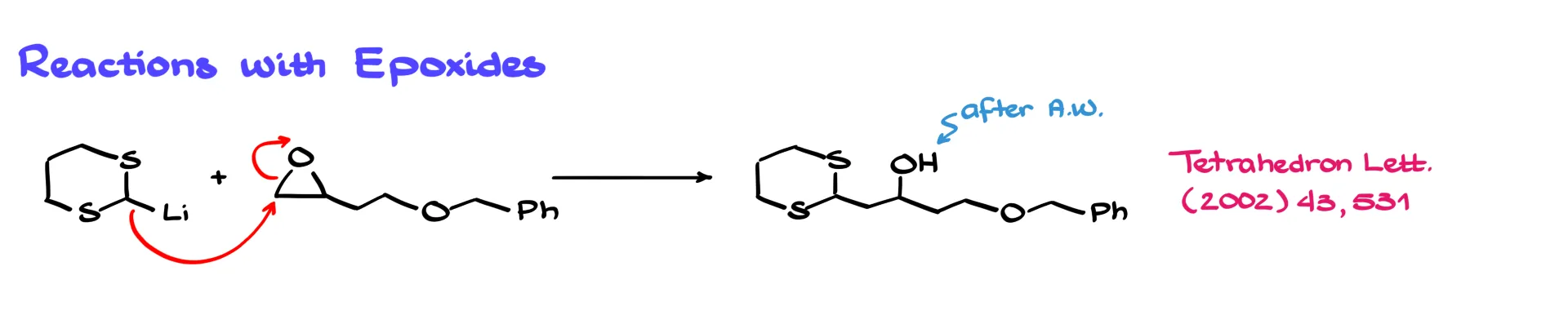
If we react our lithium derivative with an unsymmetrical epoxide, the nucleophile attacks the less substituted carbon. The epoxide ring opens, forming a new carbon–carbon bond. After acidic workup, the resulting alkoxide is protonated and we obtain the alcohol product.
Epoxides can also be used in a clever way to generate cyclopropanes.
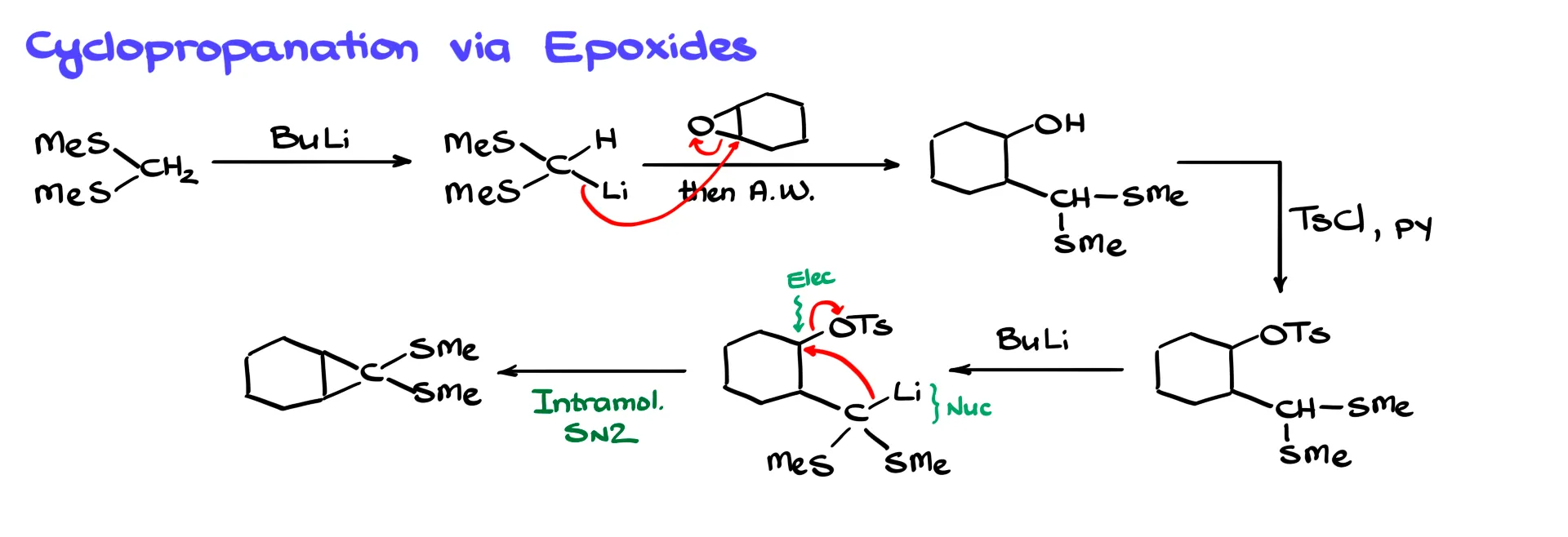
Imagine starting with a simple thioacetal. Treating it with butyllithium forms the lithium derivative. If we react that with an epoxide derived from cyclohexane, the nucleophile opens the epoxide and forms a new carbon–carbon bond. After acidic workup we obtain a product containing a newly formed OH group.
We can convert that OH group into a good leaving group using tosyl chloride, forming a tosylate. If we treat the molecule again with butyllithium, the hydrogen between the two sulfurs can still be removed, generating another lithium derivative.
Now we have a nucleophile and an electrophile in the same molecule. They react intramolecularly through an SN2 reaction. The tosylate leaves and a new carbon–carbon bond forms, producing a cyclopropane ring.
This epoxide-based strategy for cyclopropane formation is commonly used in total synthesis.
Reactions with Aldehydes and Ketones
Another class of electrophiles is aldehydes and ketones.
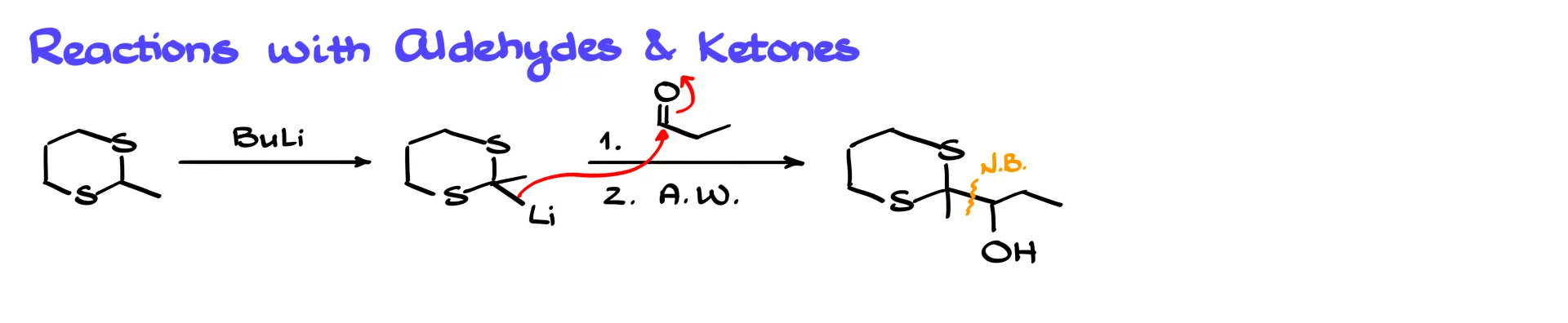
We convert the thioacetal into its lithium derivative and react it with an aldehyde, such as propanal. The nucleophile attacks the carbonyl carbon, forming a new carbon–carbon bond. After acidic workup we obtain the corresponding alcohol.
Acylation Reactions
Acylation reactions are also possible and commonly used.
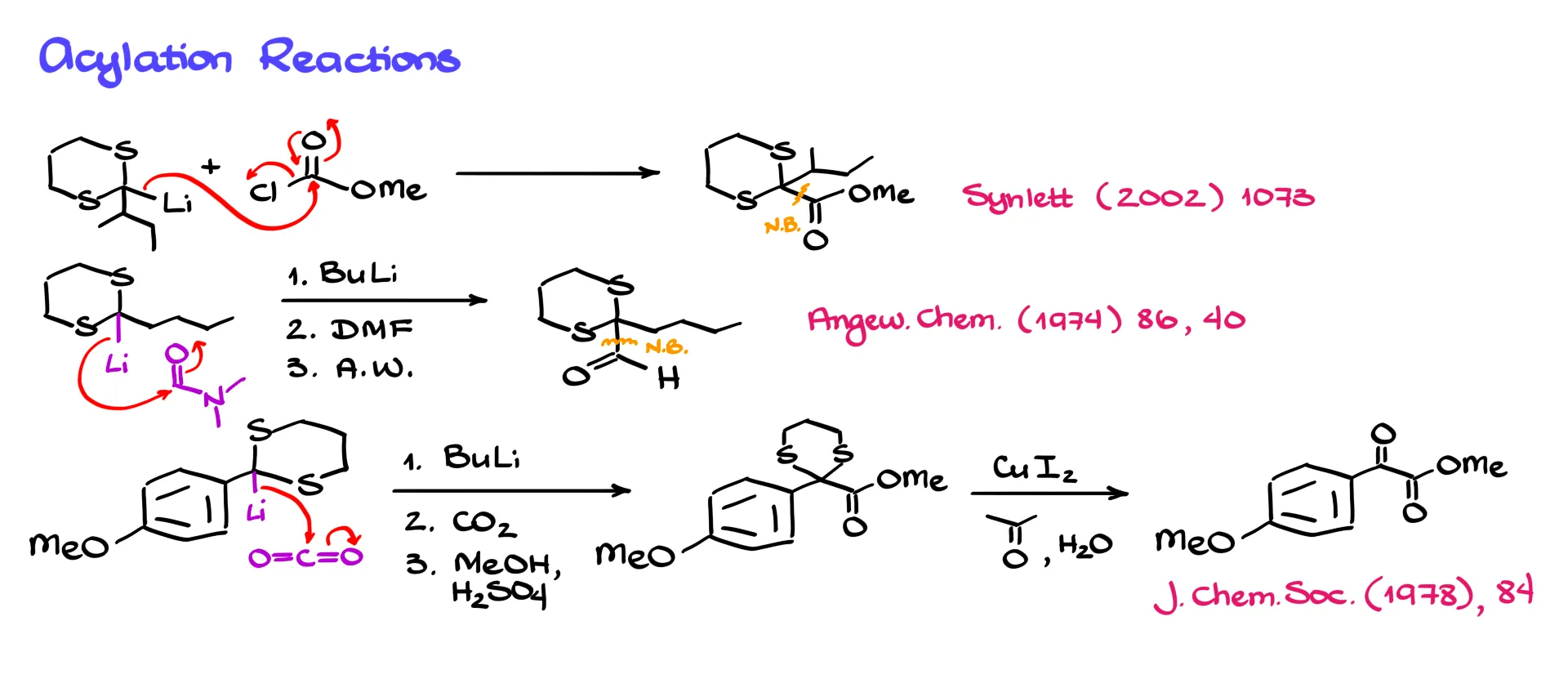
For example, the lithium derivative can react with an acid chloride. The nucleophile attacks the carbonyl carbon, electrons move up to oxygen and then back down as chloride leaves, forming a new carbon–carbon bond.
Another useful reagent is dimethylformamide, DMF. After forming the lithium derivative, we react it with DMF. The nucleophile attacks the carbonyl carbon of DMF, and during acidic workup the nitrogen-containing group is removed. The result is formation of a new aldehyde.
Similarly, reaction with CO₂ allows us to introduce a carboxyl group. The nucleophile attacks carbon dioxide, electrons shift onto oxygen, and after workup we obtain a carboxylic acid. If we then perform a Fischer esterification, we obtain the corresponding ester. Finally, hydrolysis of the thioacetal yields a useful dicarbonyl compound.
As you can see, reactions with carbonyl-type electrophiles are extremely versatile.
Michael-Style Conjugate Addition
Another possible reaction is Michael-type conjugate addition, although the literature is somewhat controversial on this point. Some reports suggest the reaction does not work well, while others show successful examples.
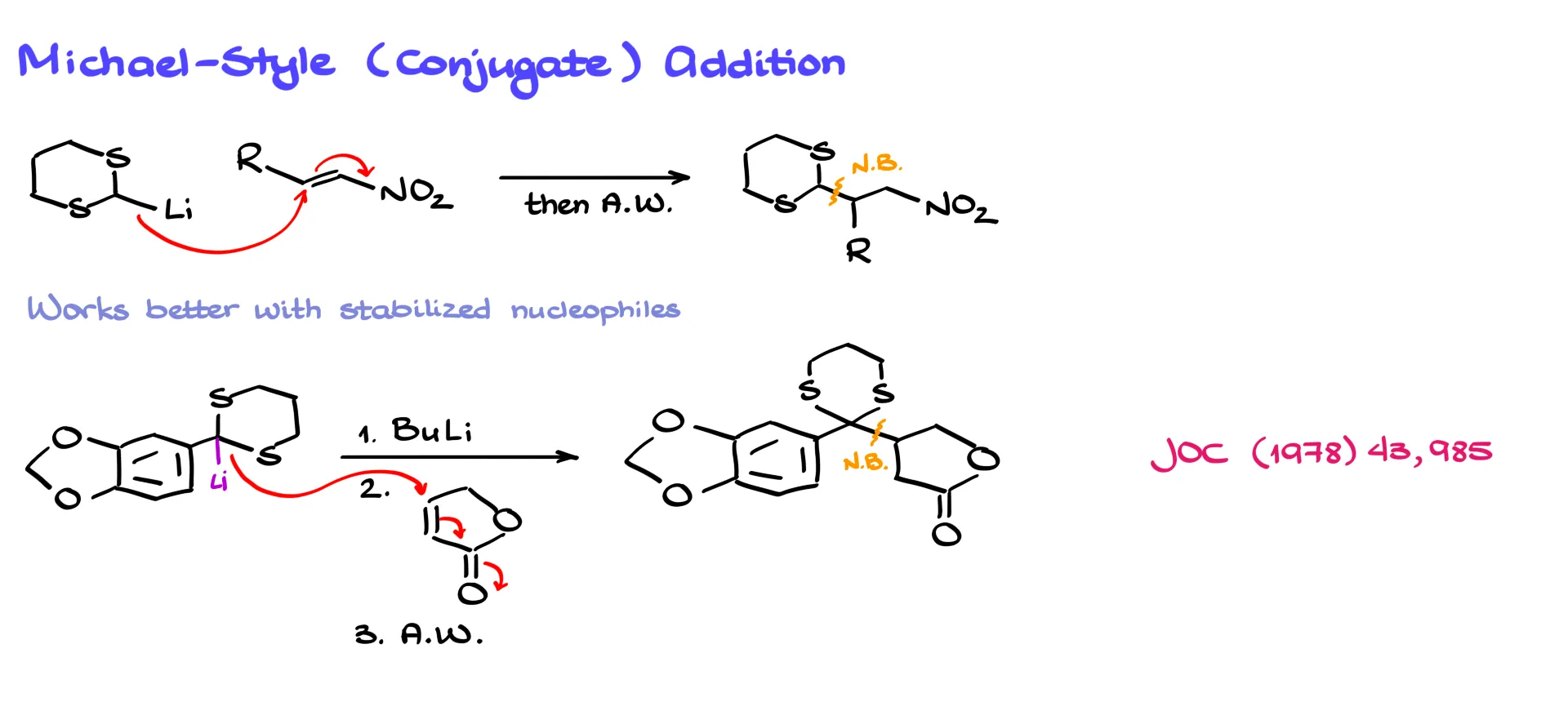
It appears to work best in two situations. The first is when the electron-withdrawing group on the alkene is something like a nitro group rather than a typical carbonyl. In that case a normal conjugate addition can occur.
The second situation is when the lithium derivative is resonance stabilized. For example, if the negative charge can be delocalized into a phenyl ring, the reagent behaves more like a classical Michael donor. In that case the nucleophile attacks the β position of the α,β-unsaturated system and forms the conjugate addition product after acidic workup.
Aromatic Substitution and Addition
Finally, lithium derivatives of thioacetals can also participate in nucleophilic aromatic substitution. Nitro-substituted aromatic rings generally do not work well because nitro groups can undergo unwanted electron transfer with organolithium reagents. However, the reaction works better with electron-deficient systems such as pyridine derivatives or aromatic rings complexed with chromium.
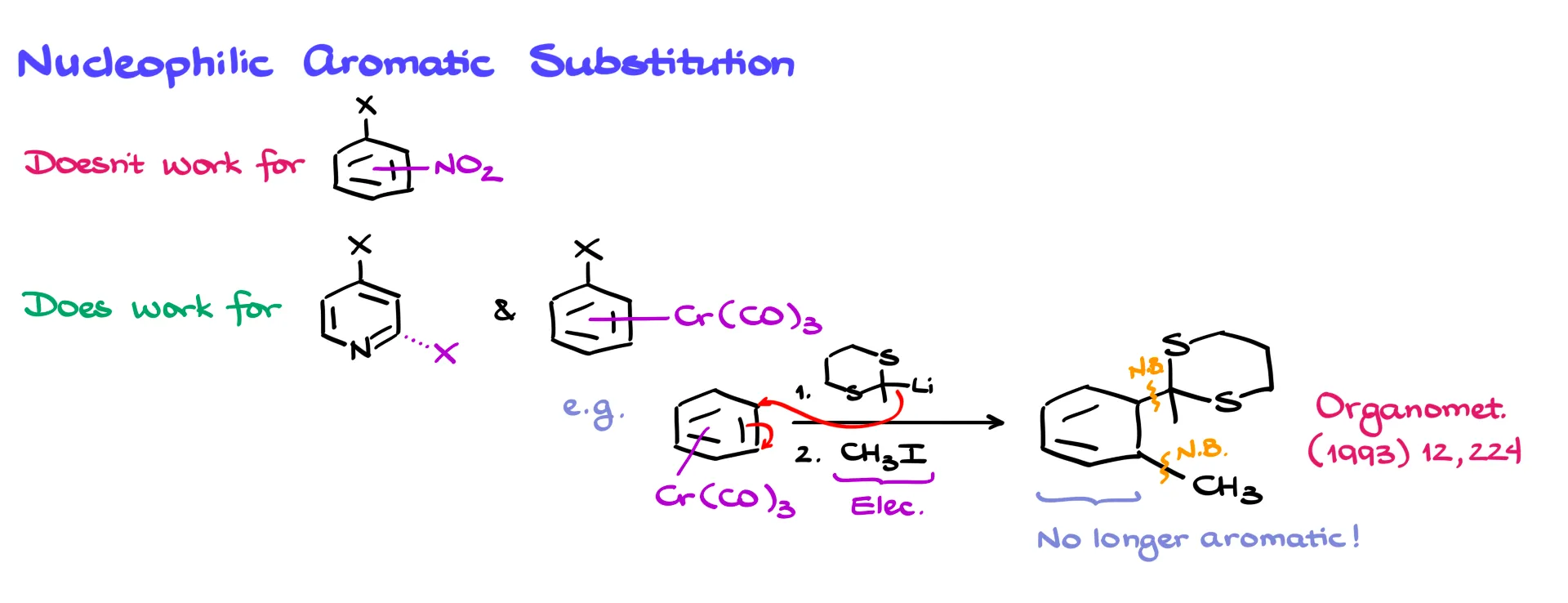
In such cases the nucleophile adds to the aromatic ring, forming a new nucleophilic intermediate that can then react with an electrophile such as methyl iodide. This sequence forms two new carbon–carbon bonds and destroys aromaticity, generating a 1,3-cyclohexadiene system that can be further modified.
Mozingo Reduction
After performing the desired transformations with our lithium derivatives, we usually want to remove the sulfur atoms.
One method is Raney nickel desulfurization. Raney nickel is a specially prepared nickel catalyst that can remove sulfur from molecules such as sulfides, thiols, and thioacetals. In the process it forms nickel sulfide and recombines the remaining alkyl fragments.
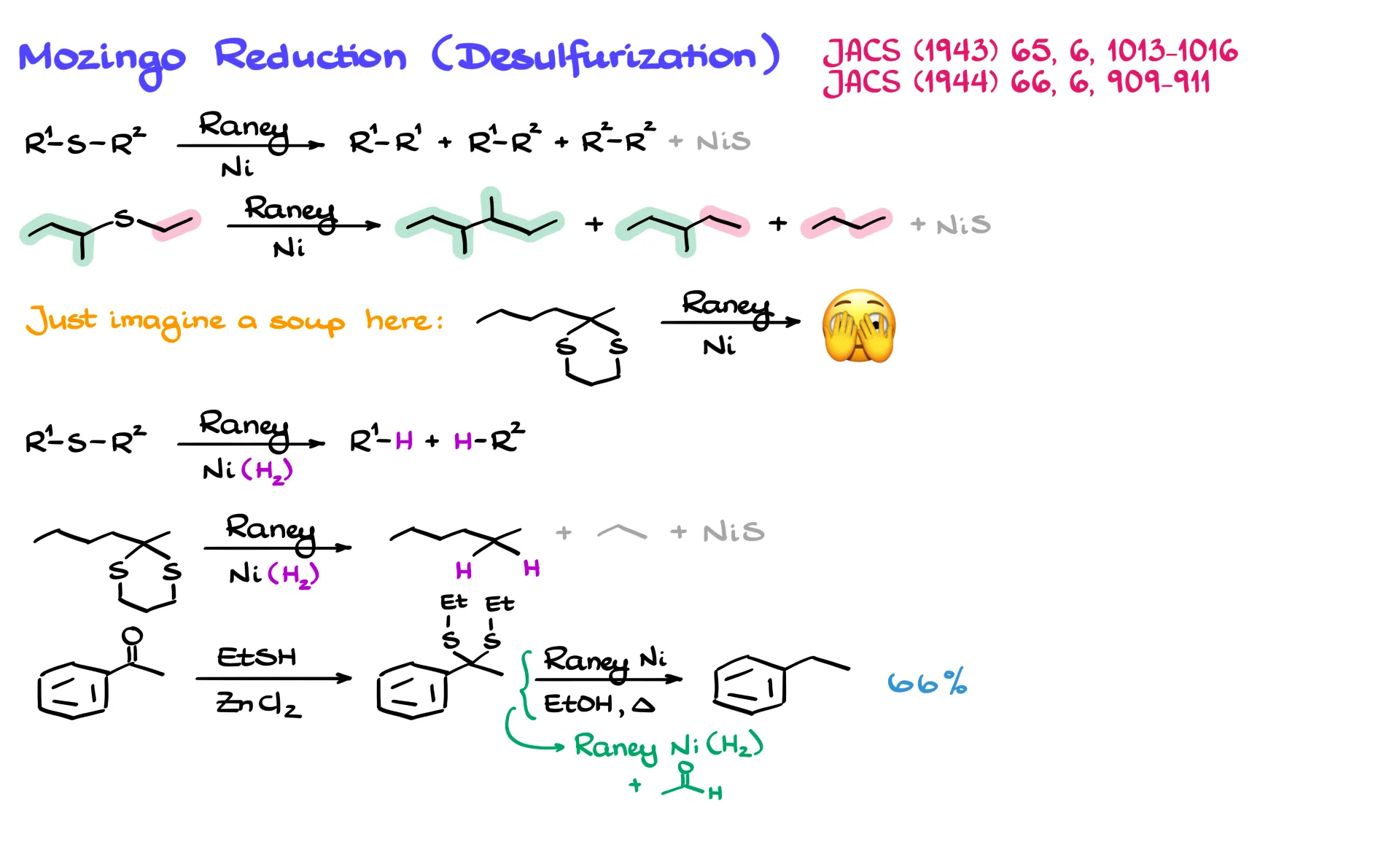
If hydrogen is present, either externally or adsorbed on the surface of the Raney nickel, the reaction instead produces the corresponding alkanes. In the case of a thioacetal, this means the carbonyl carbon is reduced to a CH₂ group.
For example, starting with acetophenone, we can react it with a thiol in the presence of zinc chloride to form the thioacetal. Treating that compound with Raney nickel in ethanol removes the sulfur atoms and reduces the carbonyl carbon to CH₂, producing ethylbenzene.
An interesting detail is that hydrogen does not need to be supplied externally. When Raney nickel is used in an alcohol solvent such as ethanol, the alcohol can be oxidized to acetaldehyde while releasing hydrogen, which replenishes hydrogen on the nickel surface.
Thioacetal Hydrolysis
If we do not want to remove the sulfur completely, we can instead hydrolyze the thioacetal to regenerate the carbonyl group.
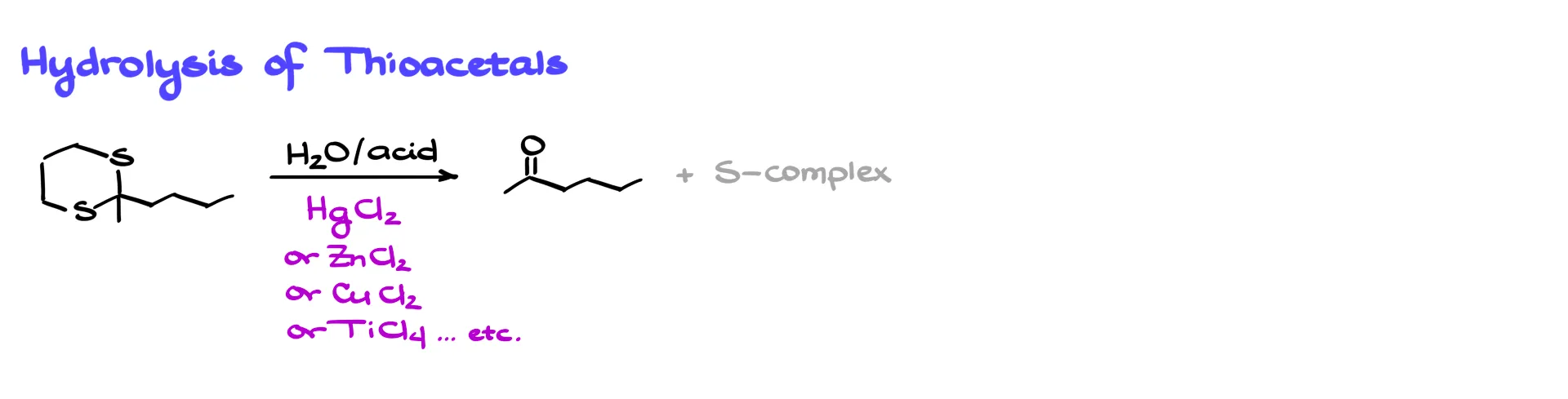
A classic method uses water and acid in the presence of mercury salts such as mercury(II) chloride. Other Lewis acids such as zinc, copper, or titanium can also work. These metals form strong complexes with sulfur, effectively removing sulfur from the equilibrium and driving the reaction toward carbonyl formation.
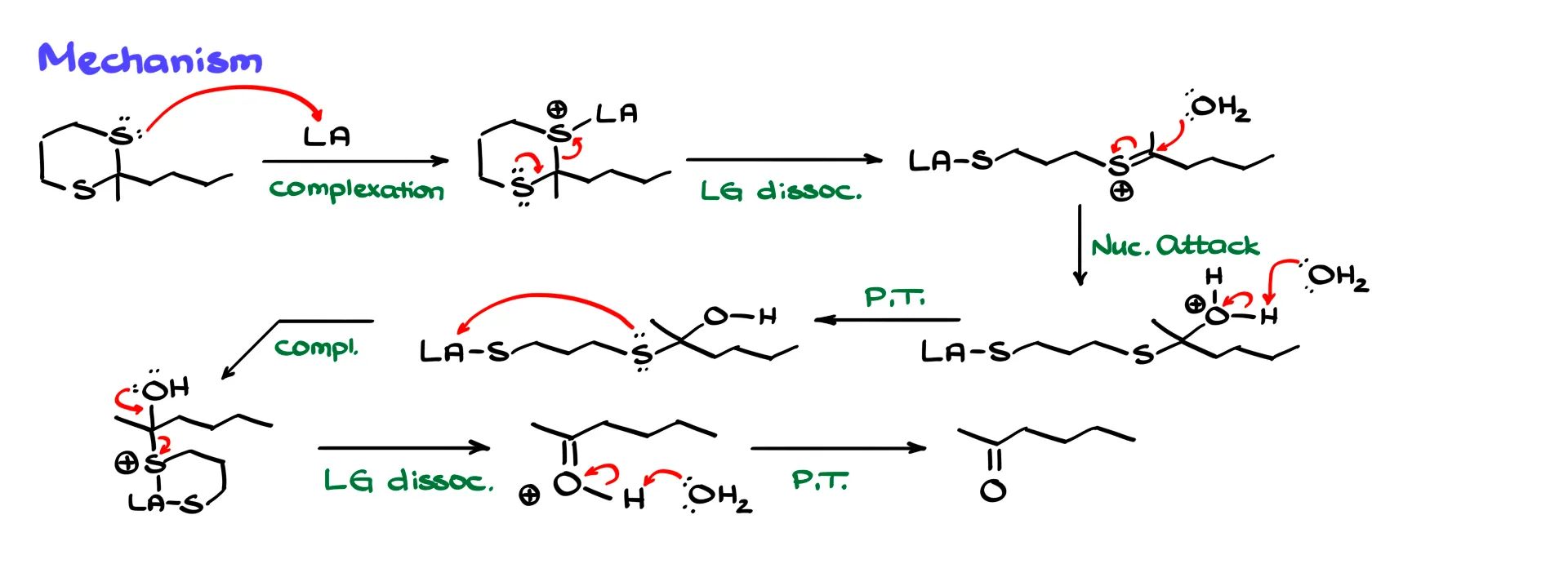
Mechanistically, sulfur first coordinates to the Lewis acid. This complex then breaks apart, generating an intermediate with a carbon–sulfur double bond. Water attacks the electrophilic carbon, forming a new intermediate. After a proton transfer and another coordination step with the Lewis acid, the sulfur-containing group leaves. A final deprotonation step releases the free carbonyl compound.
Mechanistically this process is very similar to acetal hydrolysis. The main difference is that sulfur is difficult to protonate, so Lewis acids are typically required to pull sulfur out of the system.
Now you know how to make thioacetals, how to perform reactions with their lithium derivatives, and how to remove them once they have served their purpose.