Alkynes
Alkynes have a very similar reactivity to alkenes. They undergo the electrophilic additions like halogenation and hydrohalogenation. They can also be reduced with the aid of a heterogeneous catalyst or oxidized via several techniques. Alkynes do, however, have a number of unique reactions that you’re not going to see with other functional groups.
Catalytic Hydrogenation of Alkynes
In this reaction you’re adding one or two equivalents of hydrogen to the alkyne reducing it to either an alkene or an alkyne depending on the exact conditions of this reaction.
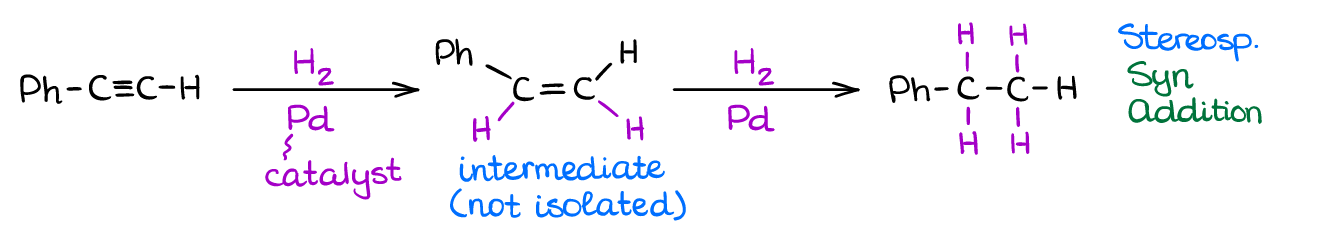
If you use your typical heterogeneous catalyst like Ni, Pt, or Pd, you’re going to have an exhaustive hydrogenation. A complete hydrogenation results in the addition of two equivalents of hydrogen gas to the molecule yielding an alkane product.
However, when you use a “poisoned” catalyst, you’re going to see only a partial reduction to alkenes. The “poisoned” catalyst (sometimes referred to as Lindlar’s Catalyst) is Pd that has been deactivated with lead or barium salts and quinoline. Like in the case of alkenes, the catalytic addition of hydrogen to alkynes is a syn process giving a cis-product.
Nucleophilic Reactions of Acetylenic Anions
Terminal alkynes are a somewhat acidic and can be deprotonated. Since the pKa value of terminal alkynes is about 25, they do require a very strong base to deprotonate them. A typical example of such a base is sodium amide NaNH2.
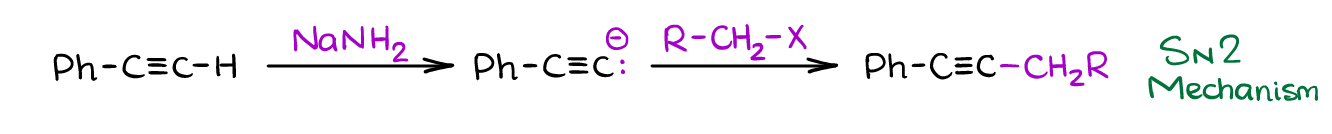
Once you form the acetylenic (or alkynyl if you like to call it this way) anion, you can react it with a wide range of compounds. The most common reaction you’re going to see in your course is a reaction with primary alkyl halides. That’s a powerful reaction because it gives an easy access to making a new carbon-carbon bond. Plus, you’ll still have an alkyne functional group that you can then modify to fit your synthetic needs.
Partial Reduction of Alkynes with Sodium in Liquid Ammonia
We have already seen a little earlier that alkynes can be partially reduced to give an alkene using the Lindlar’s catalyst. That reaction gave us a cis-alkene. But what if we need a trans-alkene? Well, there’s a reaction that can accomplish just that!
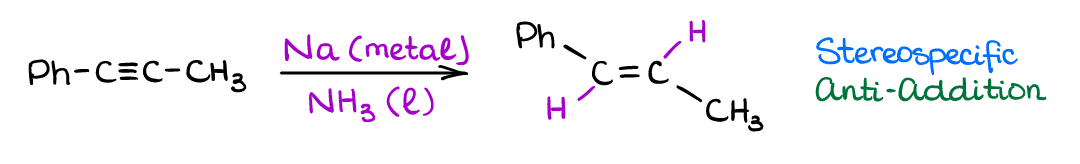
This reaction uses (most commonly) sodium metal (Na) as a reducing agent. You may also occasionally see lithium (Li) used in the same way. The mechanism of this reaction is rather complicated and involves several electron transfer steps giving exotic anion-radical species. You may or may not need to know the mechanism of the reaction for the test, however, you will be responsible for the reaction outcome and the reaction stereochemistry (trans-alkene product).
Hydration of Alkynes
Water can add to alkynes in acidic conditions like the same reaction in alkenes. Alkynes, however, are much less reactive towards any electrophiles than alkenes. Therefore, this reaction will require very harsh conditions, vigorous reflux, and extended reaction times.
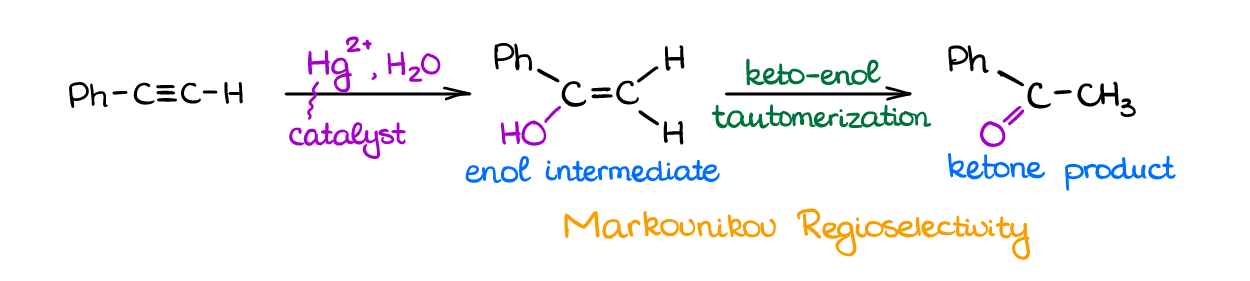
To make this reaction easier, we’re going to use the Hg2+ for the catalyst. The addition of mercury makes a more stable reactive intermediate lowering activation energy and making reaction faster and more efficient.
Reaction has Markovnikov regioselectivity and forms and enol intermediate. The intermediate is not stable though and rearranges into a more thermodynamically stable ketone product via keto-enol tautomerization.
Hydroboration-Oxidation of Alkynes
This is another reaction similar to what you already know for alkenes. In this reaction, an alkyne reacts with a borane in the first step and then you oxidize the resulting intermediate with hydrogen peroxide in basic media.
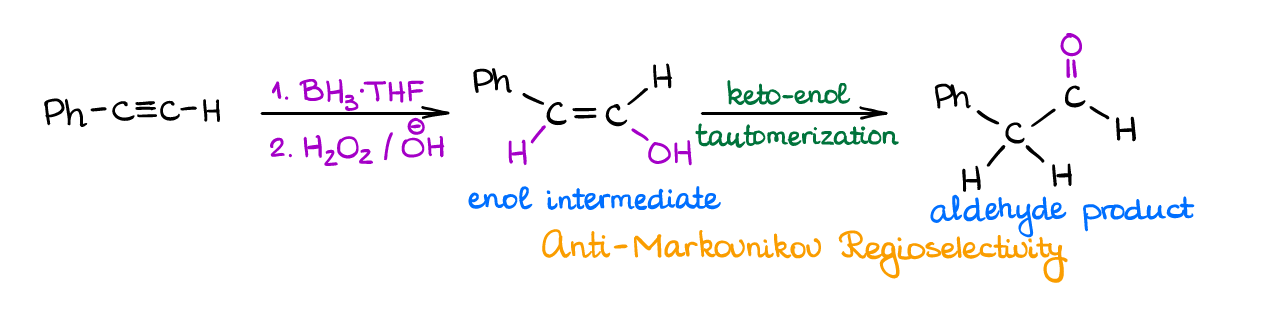
This reaction shows the anti-Markovnikov regioselectivity and produces an enol intermediate which quickly tautomerizes into a corresponding carbonyl. Reaction gives an aldehyde when you preform it with a terminal alkyne, and a ketone when you perform it with an internal alkyne.
In the case of internal alkynes, you can perform hydroboration-oxidation in a very regioselective way by using a bulky borane like 9-BBN or similar compounds. The steric hindrance that a bulky borane experiences in this reaction will drive reaction towards the product with oxygen on the less sterically hindered atom.
Ozonolysis of Alkynes
This is a useful reaction for cleaving triple bonds. Unlike its analogous reaction of alkenes, we only have one way of performing this one and it doesn’t require a workup step.
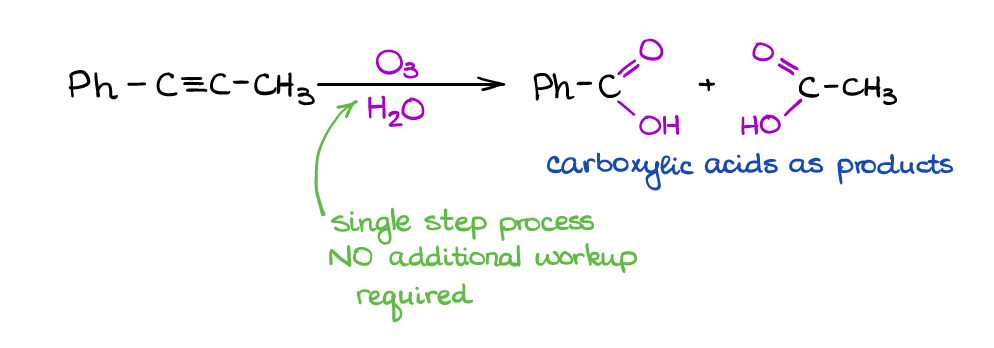
The mechanism of this reaction is something that organic chemists still debate about. You won’t need to know the mechanism for the exam (at least, I’ve never seen it on the exam in all the years of me working with students). However, you will be responsible for the outcome, which is a pair of carboxylic acids.
Halogenation of Alkynes
This is a typical electrophilic addition to alkynes similar to the same reaction of alkenes. Since alkynes have two π-bonds, you can have two equivalents of a halogen adding to the triple bond.
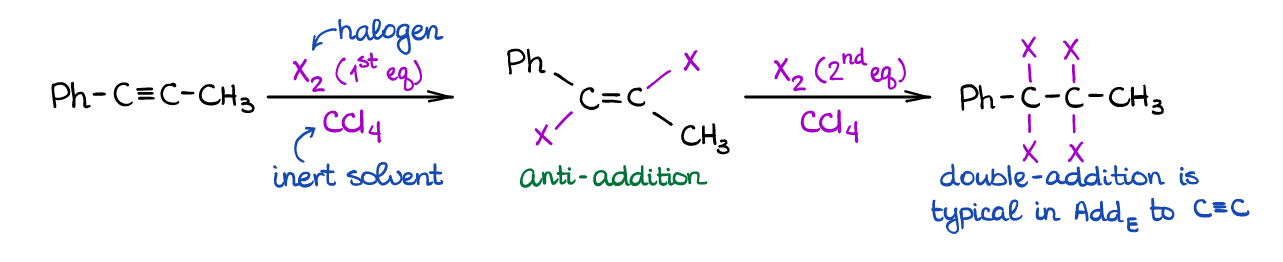
Noticeably, the first equivalent of the halogen adds in a very stereospecific way (anti-addition) just like with alkenes. Also, since alkenes are generally more reactive in electrophilic additions, the addition of the second equivalent happens quite easily. The ease of the second equivalent addition makes it somewhat challenging to “catch” the product of the single equivalent addition, however, it’s still doable.
Hydroxyhalogenation of Alkynes
When you react an alkyne with a halogen in a large access of water, you will see the competition between the H2O and Br– for the place of a nucleophile in this reaction. For as long as you have a large excess of water, it generally “wins” ang gives a corresponding halohydrin intermediate.
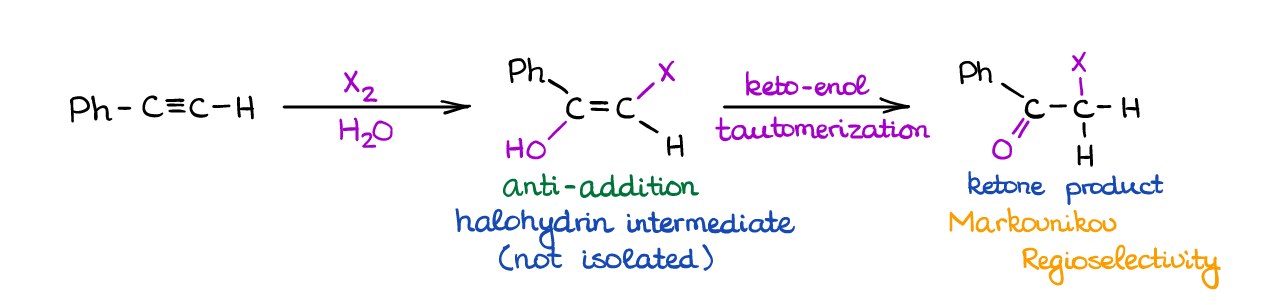
Since the halohydrin is also an enol like in the previous reactions, you’re going to see a rapid keto-enol tautomerization giving a halogenated ketone as a final product. The initial addition is stereospecific (anti-addition) giving you a trans-product.
Alkoxyhalogenation of Alkynes
Like hydroxyhalogenation above, this reaction uses a large excess of an alcohol to act as a nucleophile.
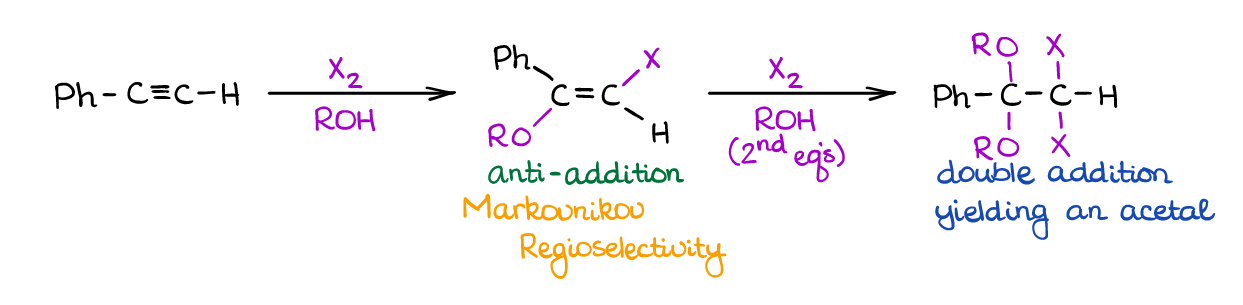
Reaction is on the more exotic end of a spectrum and has little synthetic applicability. Additionally, it suffers from the double-addition and poor regioselectivity of the second addition. Nonetheless, versions of alkoxyhalogenation of alkynes did find some success in a few synthetic procedures out there. I’ve also seen this type of reaction of alkynes on the tests as a “curved ball” or extra points question, so it may still be a worthwhile idea to keep in on the backburner.
Hydrohalogenation of Alkynes
If you were to ask me the “classic” reaction of alkenes and alkynes, that, undoubtedly, be the hydrohalogenation of alkynes. The reaction of a π-bond with HX is, perhaps, one of the first reactions that you’re going to see in both sections on alkenes and alkynes in your course.
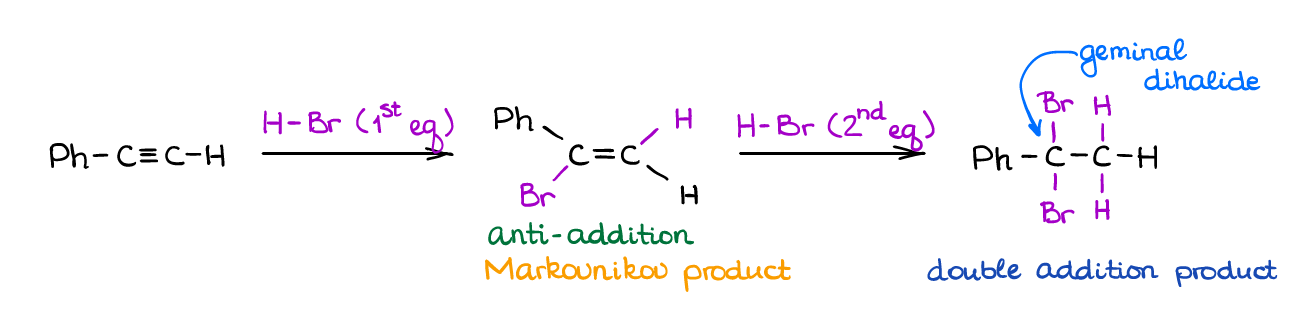
The reaction has a strict Markovnikov regioselectivity for both addition steps. It also shows the stereospecificity giving an anti-addition product (trans-product) in the first step. Like in all reactions of alkynes, it’s difficult to “catch” the product of the first addition but it’s still possible so you can feel free to use the “1 equivalent” reactions on your test and in the synthetic procedures.
Free-Radical Hydrohalogenation of Alkynes
When you need an anti-Markovnikov addition product to a π-bond, peroxides are your friends! The free-radical mechanism of the addition gives you an anti-Markovnikov product and is a useful synthetic reaction for the simple molecules.
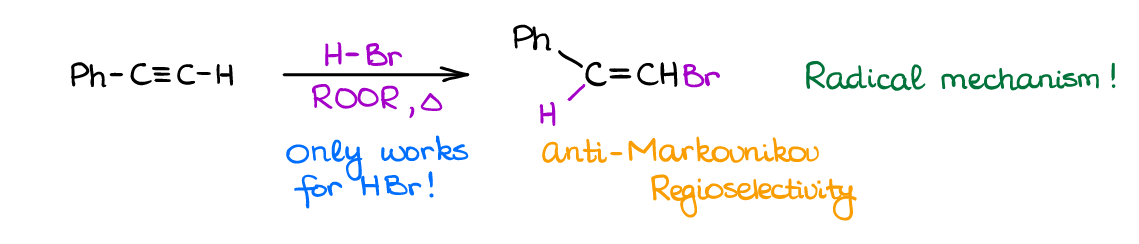
The complication you’re going to see with this reaction is a strong tendency to give polymerization reaction severely lowering the yields. However, for our purposes, we can assume we can stop the unwanted side products, so again, feel free to use this reaction on the exam when working on the multistep synthesis. Importantly though, this reaction has very poor stereoselectivity, so you are going to get the cis– and trans-products as a mixture.