Multiple Directing Effects and Introduction to Multistep Synthesis
In this tutorial, I want to talk about the electrophilic aromatic substitution in multisubstituted aromatic compounds and planning of the multistep synthesis of aromatic compounds. Let’s start by looking at this reaction.
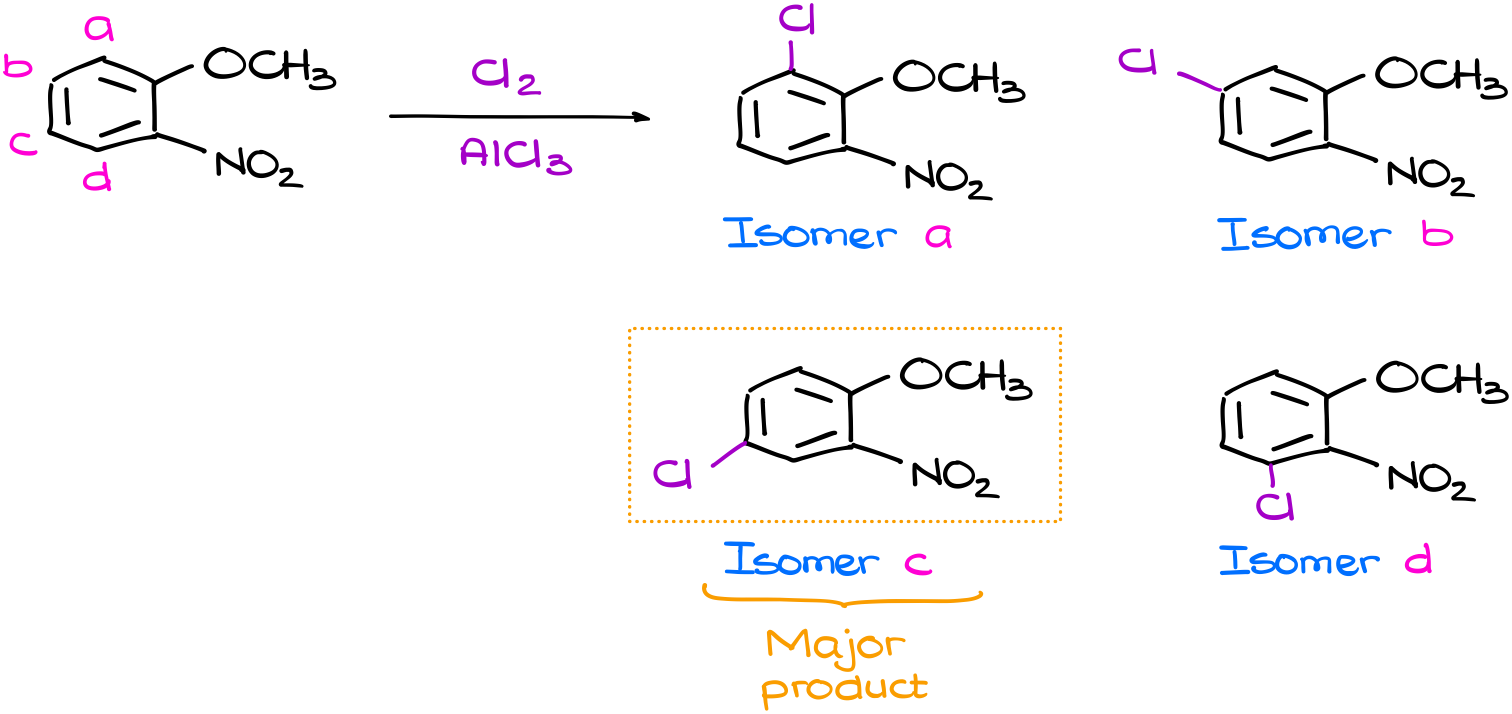
As our molecule is not symmetrical, there are four possible places where we can perform the substitution. This gives us four possible products. Now, the question is: which one is going to be our major product? To answer that question, you’d probably need to draw the resonance structures for all the intermediates (σ-complexes) and go from there. But what if I told you, that no, you don’t actually need to do that. What if I told you that you can tell the major product and skip the tedious resonance? Like, in this case, the isomer C is going to be my major product. And I could figure out by doing a very simple analysis of the starting material.
Ok, enough of this suspense!
Directing Effect Shortcut
There’s a very simple shortcut. Let’s start by looking at an aromatic compound with an Electron-Donating Group (EDG).

The electron-donating groups are activating groups and they bring an extra electron density to the ring. Specifically, they make the o/p-positions more electron rich. Thus, the electrophile is going to be more likely to attack those positions. We also know that in the case of the o/p-directors, the para-position is typically favored. So, if this position is available, we’re going to attack that position.
In the case of the Electron-Withdrawing Groups (EWG), we have the opposite picture. The electron-withdrawing groups will pull the electron density out of the ring making it more positive. The electron-withdrawing groups will put the partial positive charges on the o/p-positions deactivating them.
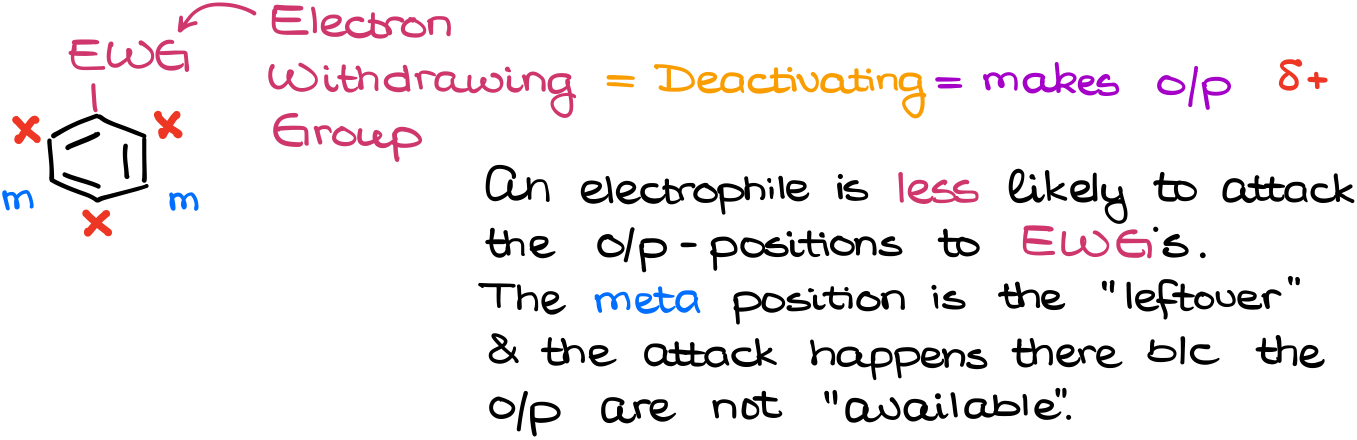
Thus, the electrophile will be less likely to attack those positions. I personally really don’t like the term “meta-director.” Unlike the donating groups which make the o/p-positions more attractive for electrophiles, the electron-withdrawing groups are not actually directing the substitution into the meta position. The deactivating groups make the o/p-positions less attractive for electrophiles instead. The meta position is nothing more but a leftover that was not deactivated.
So, I prefer to think about the activating groups as the o/p-activators while the deactivating groups are the o/p-deactivators.
Examples of Multiple Directing Effects
For instance, let’s look at this example.

If we analyze it from the perspective of the o/p-activating and deactivating effects, we’ll see that the methoxy group (in green) is the activating group. Thus, it’s going to activate its own o/p-positions. At the same time, the nitro group (in pink) is the deactivating group. So, it’s going to be deactivating its own o/p positions. I like to use the arrows and crosses on my molecule. This helps me to easily map the possible substitution positions in my compound. So here, I have a couple of positions that are likely candidates for the substitution, and a couple of positions that are not.
This analysis gives us two possible products. And as we know that the para-product is typically the one that forms in a larger quantity due to the steric factors. So, we’ll assign the major product to the first molecule here.
Here’s another example.
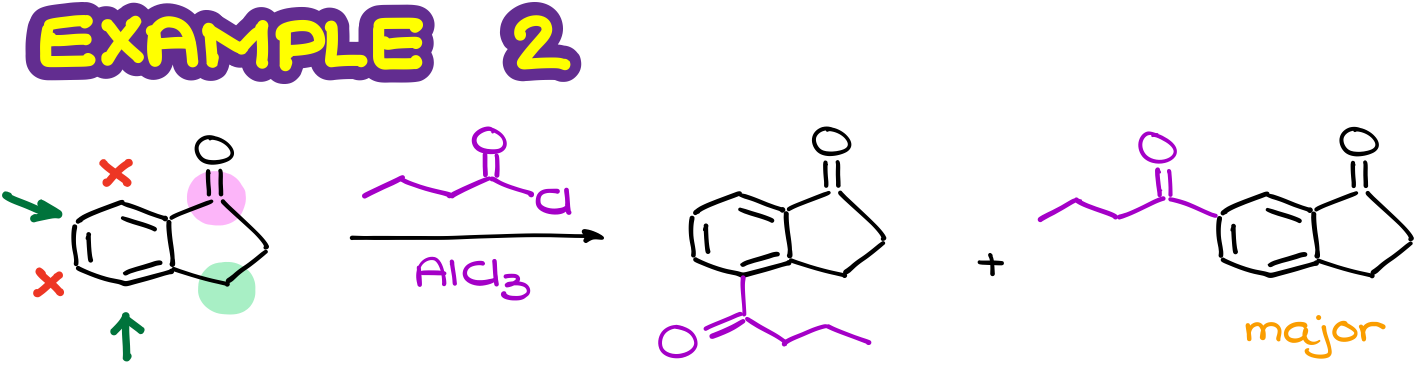
By doing the same analysis as above, I can quickly map the activated positions. Those are going to be the positions that are the o/p to my activating group. Notice that here I only pay attention to the first atom attached to my ring as it exhibits the most influence. So, the green CH2 group here is the weakly activating and electron-donating group. And it doesn’t really matter that a few atoms down the chin it is connected to a carbonyl. That carbonyl is too far to matter for the CH2 group.
Likewise, I have a pink carbonyl that I’ll treat as a moderately deactivating group. And again, the fact that it is later connected to other stuff is pretty much irrelevant. So, I can quickly map the deactivated positions and proceed with drawing of my products. And here again, due to the steric factors, I’ll choose the molecule to the right as my major product.
Conflicting Directing Effects
Now, in all previous examples my groups were always playing along just fine. We haven’t seen any conflicts or ambiguity. This is, of course, not going to be the case every single time.
So, what happens when the groups fight with each other? Like for instance in this reaction, where I have a weakly activating group activating its own o-positions. And I also have a strongly activating group which activates its own o-positions.
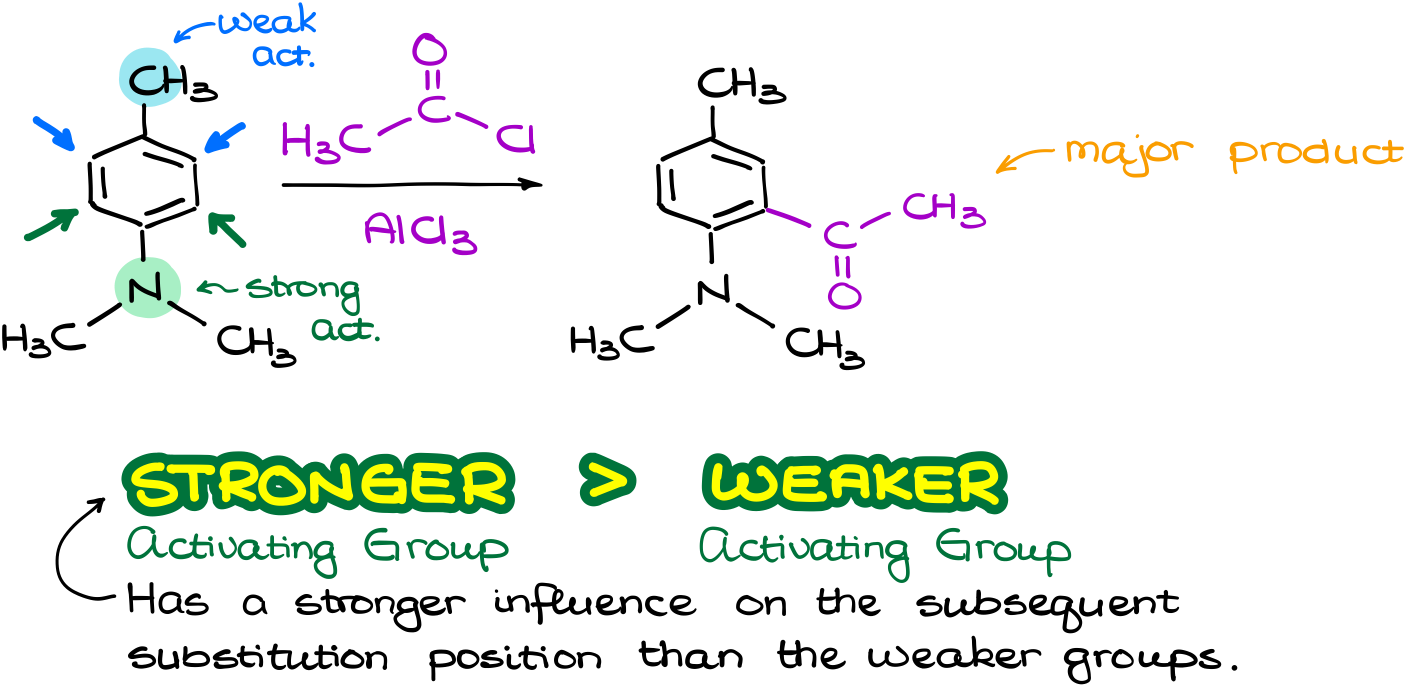
So, what are we going to do in this case? Draw the resonance structures to help us decide? Hell no! That takes too much time. Luckily, we can just remember that the stronger activating group will typically overpower the weaker activating group. Don’t discount the sterics though. If your strongest activating group is very bulky, then attacking the o-position next to it may be very difficult. But for as long as we don’t have anything extreme, the electronic effects will be more important. So, in this case the major product will be the one where we add a new group to the o-position to the strongest activating group, which is our dimethylamino group.
We can also have a situation in which we have a conflict between the activating group and a deactivating group that affect the same positions in the molecule. In cases like this one, typically, the activating group overpowers the deactivating group. So, the substitution is going to go into one of the positions that the activating group is directing us to.

Predicting the major product in these reactions is a little more challenging though. One thing that we can say for sure is that the substitution is unlikely to occur in between the groups due to the steric hindrance. But which of the two other products is going to be major here? Well, that’s where the challenge is. You see, different textbooks have slightly different criteria for this case. Some prioritize the p-position to the donating group making the molecule to the right major. Some prioritize the distance from the electron-withdrawing group, the further the better. This would make the left molecule major. The experimental evidence suggests that the former is more important in the case of strongly activating groups. And the latter is more important for the weakly activating groups. So, I’ll leave it with you and your instructor since, at the end of the day, it’s them who’s going to give you your grade. But I personally will side with the experimental evidence.
Now, remember how I’ve mentioned earlier that it’s a good idea to think about the m-directors as o/p-deactivators instead? Well, that’s because the molecule can be completely deactivated. Like, for instance, in this case.

Here I have the pink nitro group deactivating its positions. And I have the blue nitro group deactivating its own positions. So, as the result, the entire molecule is so deactivated that for all intents and purposes, it’s too unreactive in the electrophilic aromatic substitution. It doesn’t mean that the reaction is entirely impossible. No, it is quite possible. But remember that the deactivating groups make reaction go slow. So, when the entire molecule is deactivated, the reaction is going to be SO slow, that for any practical purposes it’s not going to be feasible at all.
Introduction to the Multistep Synthesis of Multisubstituted Aromatic Compounds
Now, when we know how to quickly approach the directing effects in electrophilic aromatic substitution reactions, let’s talk about the multistep synthesis. Here I have a typical exam question.

When it comes to questions like that, I suggest you first map out the groups that you’ve added to your compound and the reactions you’d need to know to add them.

In this case, we’re going to be adding a halogen, chlorine. This can be done via a simple halogenation reaction. We’re also going to be adding the ketone via the Friedel-Crafts acylation reaction.
When I’m doing this type of analysis, I’m also noticing the nature of my groups and whether they are activating or deactivating groups. This part is very important for planning out your synthesis.
And here’s why. If I don’t pay attention to the directing effects of my groups and, for instance, do the halogenation first I’ll end up with the weakly deactivating BUT the o/p-directing group on my ring first.
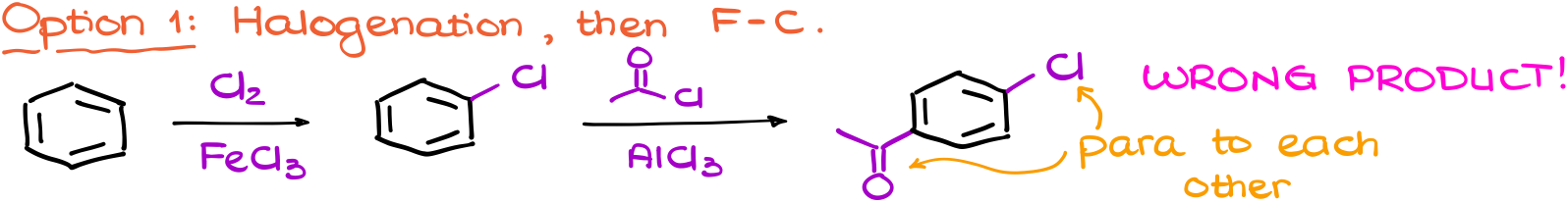
This means that I’ll end up with the wrong product after my Friedel-Crafts acylation reaction.
However, if I perform my steps in the reverse order, I’ll first have the meta-directing group on the ring. This means that my halogenation going to add the chlorine atom to the meta position where we want it to be.
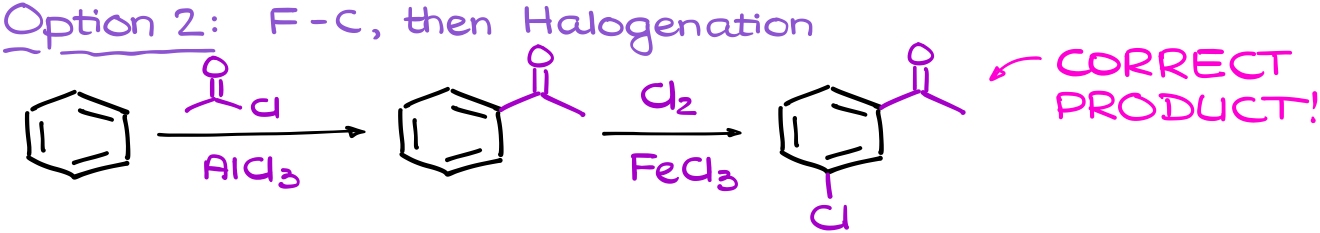
At the beginning, the multistep synthesis is going to be a bit of a trial and error for you. But that’s ok. For as long as you have all the important pieced mapped out, you’ll be able to quickly go through the options and see which sequence gives you the required product. With a little practice, you’ll see that you’ll be able to map the sequence in your head and solve these questions much easier faster. So, as I always say, do a ton of practice!