Additional practice questions and answer keys are available to site members.
Sign up today or login if you’re already a member and unlock all members-only content!
SN1 Reactions with Carbocation Rearrangements
In this tutorial we’re going to continue our investigation of the SN1 reactions. However, now we’re going to pay more attention to the carbocation intermediates and their possible rearrangements. So, let’s take a look at this question. This looks like a fairly common SN1 question, so we shouldn’t really have any problems with that, right?
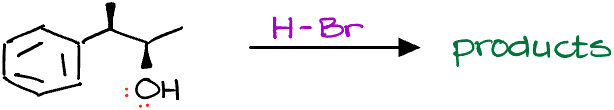
So, let me draw the mechanism really quickly and predict our products.

Based on what we’ve learned about the SN1 reactions in the last video, this should be our final product, right?
WRONG! Turns out, these are only the minor products!
So, what’s going on and where did we go wrong? If you form a carbocation intermediate, you always have to first check for possible carbocation rearrangements. The thing is, if a carbocation can rearrange to give you a more stable carbocation, it absolutely will! And since intramolecular processes are typically significantly faster than the intermolecular ones, the carbocation rearrangement will typically occur before any nucleophilic attack, or any other interactions you may think for your molecule.
Carbocation Stability Trends
So, let’s first look at the carbocation stability trend and which factors influence it.
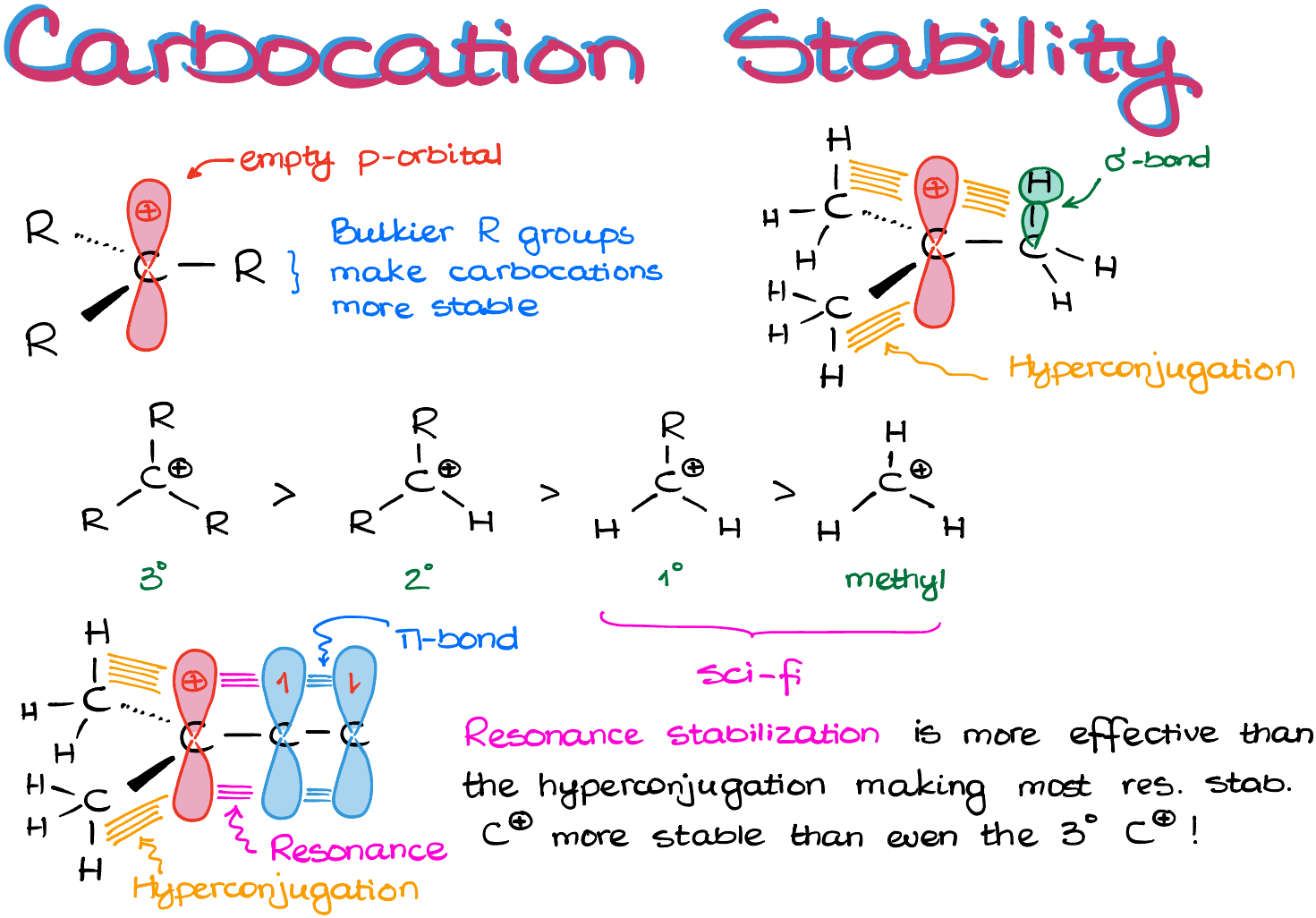
First of all, what is a carbocation?
A carbocation is an sp2-hybridized species which is trigonal planar in its geometry.
And as those species are extremely electrophilic due to the open shell nature (they have an empty p-orbital after all), they become more stable with more groups surrounding the empty orbital. So, the more “stuff” we have around our empty p-orbital, the more stable the carbocation is going to be.
Which means, we have a general trend of the 3° being more stable than the 2° which, in turn, is more stable than the 1° and the methyl carbocations.
Here’s something important though: the 1° and the methyl carbocations are so unstable, that for our purposes they are science fiction. So, never-ever-ever attempt to make a primary carbocation in an SN1 reaction unless there’s a really good reason for it. I’m not saying that the primary and methyl carbocations are completely impossible, but they are darn close to it.
Which bring me to the question: why the 3° carbocations are more stable than the 2° ones? Is there a more “scientific” explanation than “more stuff is good” explanation? And yes, there is one!
The effect we’re talking about is called hyperconjugation. In a nutshell, hyperconjugation is a partial overlap between the empty p-orbital and the nearby σ-bond on the adjacent atom.
And as the hyperconjugation is only a partial overlap, it only provides a small stabilization effect on the empty orbital. So, we wanna have multiple of those overlaps to be meaningful. Thus, the primary carbocations which will only be able to provide one hyperconjugation overlap, are still quite unstable. In the meantime, the tertiary carbocations, which have 3 hyperconjugation overlaps, are significantly more stable.
So, if the partial overlap is good, how about the complete orbital overlap? For instance, if we had a π-bond instead of a σ-bond nearby, wouldn’t it be better? And yes, it absolutely would!
If we have a full 100% overlap with a π-bond, this is going to be a resonance stabilization. And as the resonance stabilization is more effective than the hyperconjugation, most resonance-stabilized carbocations are actually more stable than even the 3° carbocations! Thus, if you can make a carbocation with resonance, always go for it as it is going to be quite stable and will form easily.
So, a quick recap: resonance-stabilized carbocations are awesome, 3° carbocations are good, 2° carbocations are ok, everything else is science fiction. Thus, we should avoid anything that’s not resonance stabilized, 3°, or 2° carbocation when coming up with the mechanism of our reaction.
Types of Carbocation Rearrangements
Cool. Now, what exactly is the carbocation rearrangement?
Carbocation rearrangements can happen via either hydride or alkyl shifts. The technical term is 1,2-hydride or 1,2-alkyl shift. We don’t need to go into details why they are called this way, just remember that if you hear one of those terms, they mean carbocation rearrangements.
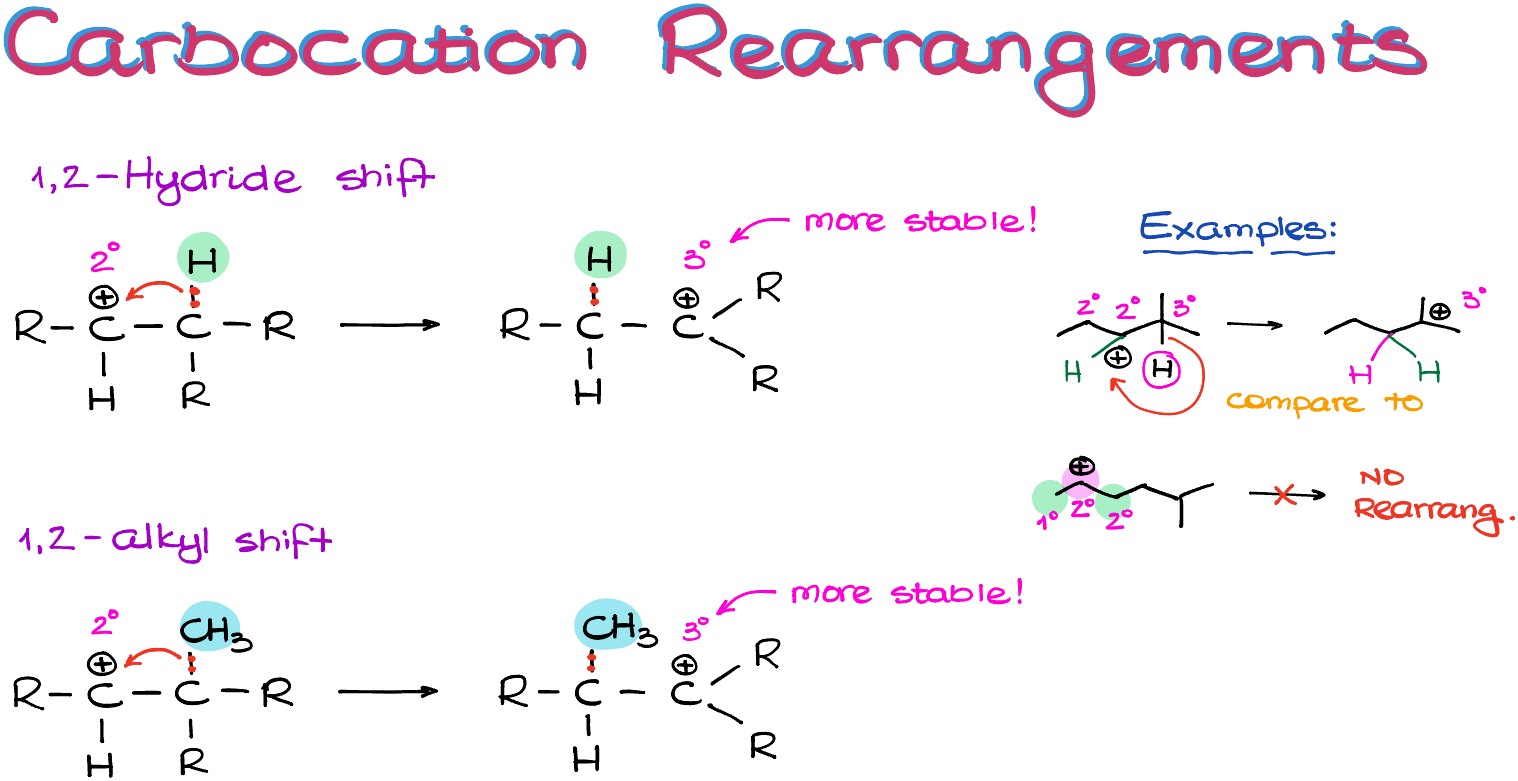
So, what actually happens, is the hydrogen atom with its electrons migrates to the atom with the empty orbital (our carbocation). Likewise, in the case of an alkyl shift, it’s the entire alkyl group with all of its electrons migrates from one atom to another.
And while it might seem like we’re moving the positive charge, especially if we’re using the skeletal structures, in reality we are not. We are moving the groups around and the new position of a carbocation is a consequence of that move. So, remember, it’s not the charge that hopping around, it’s the groups!
Ok, groups can hop from one carbocation to another. Cool beans! But why exactly does that happen?
Good question!
The driving force for the carbocation rearrangement is the carbocation stability. The carbocation rearrangement allows us to make a more stable carbocation! And since carbocations are intrinsically unstable, this is kind of a big deal.
Remember however: carbocations don’t have long-term planning skills! So they will ONLY rearrange if we have an immediate gain in stability.
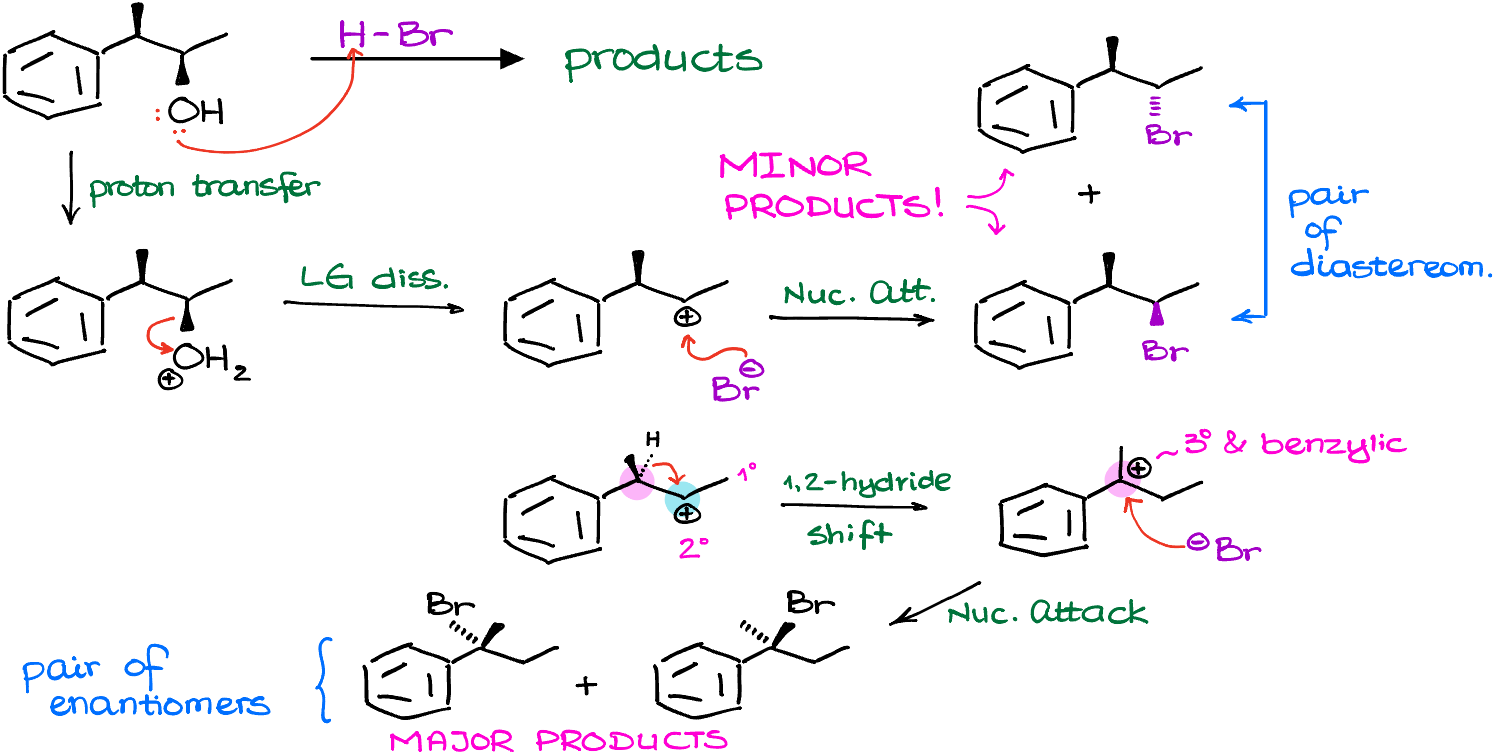
Examples of SN1 Reactions with Carbocation Rearrangements
So, for instance, here I have a couple of examples. In the first example we can immediately rearrange the carbocation via a hydride shift to make a tertiary carbocation, which is more stable than the secondary we have at the moment. In the second example, however, we cannot make a more stable carbocation immediately. And as our carbocation doesn’t “know” that after multiple rearrangements it can become tertiary, we’re not going to see any rearrangement. Remember, carbocations are not humans, they don’t have long-term planning skills. So, if you have a friend who’s lacking long-term planning skills, I officially give you a permission to start calling them “carbocation” 🤣
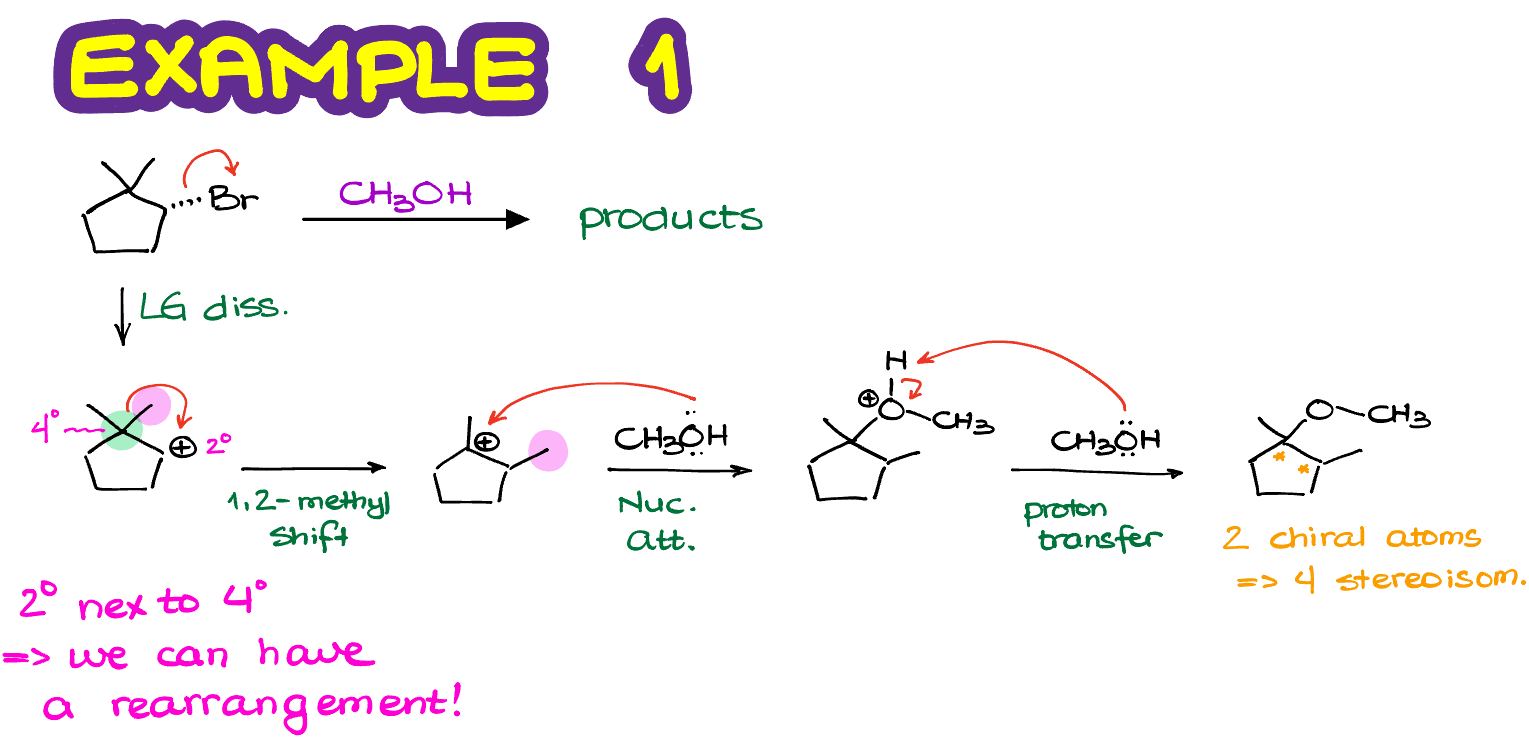
The carbocation intermediate we have in next reaction is 2°. And it is next to a primary and a tertiary carbocation. So, if we were to rearrange it to a tertiary position, we are going to immediately make a more stable carbocation.
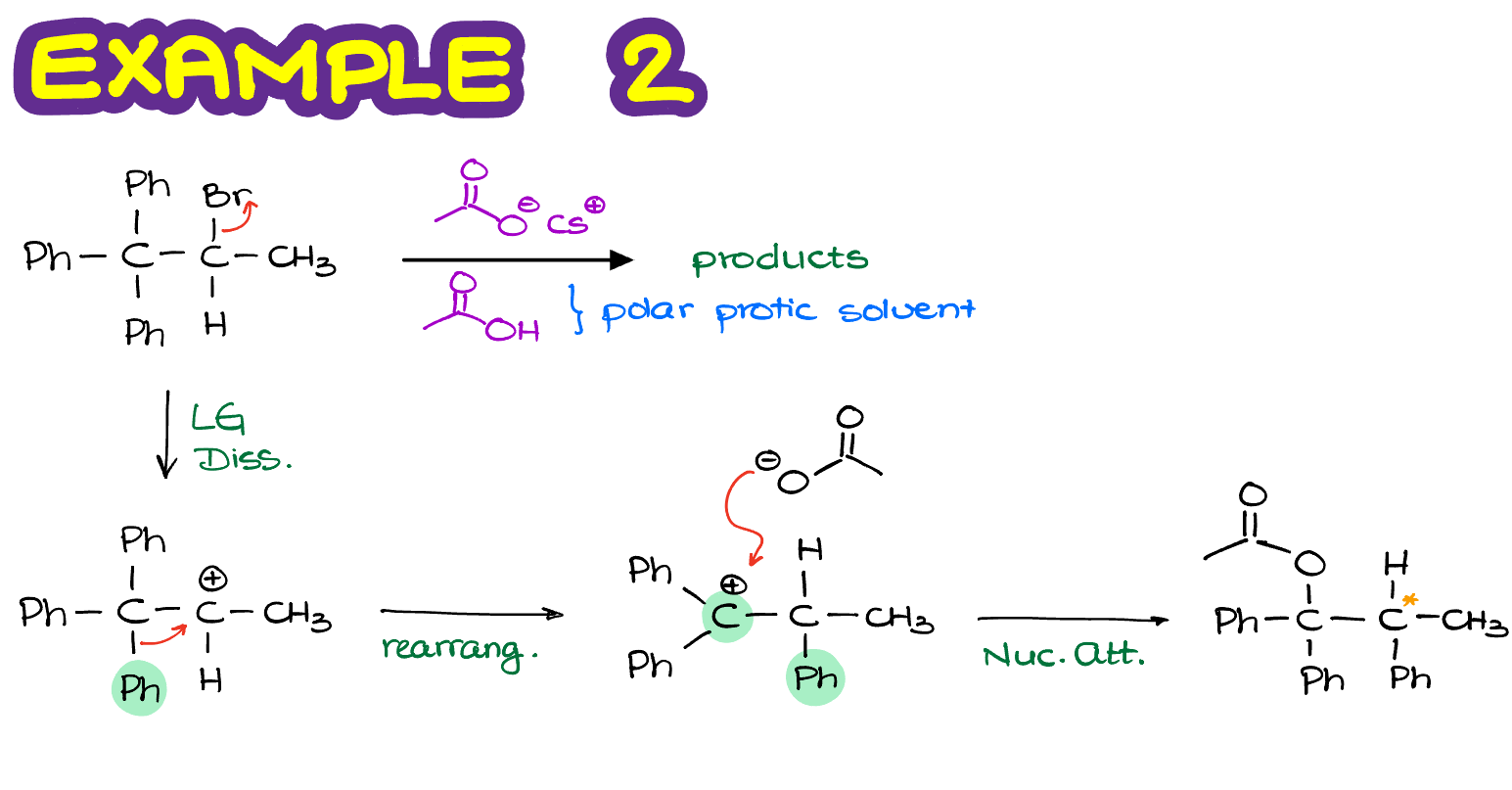
In this case, we are going to perform a rearrangement by moving the phenyl ring around and form and new 3° carbocation which is also benzylic, meaning we have a resonance stabilization from the benzene ring next to it. This carbocation is exponentially more stable than the one we’ve had to begin with. So, in this example, we are going to have the absolute majority of molecules undergo the carbocation rearrangement before the nucleophilic attack. Note: there’s an error in the figure above, there’s only 1 chiral atom the product making a pair of enantiomers.
Finally, once we perform the nucleophilic attack on our newly formed carbocation, we are going to make our actual final product.
So, remember to always check for the carbocation rearrangement in any reaction involving carbocations! If you blindly follow the steps in the mechanism and don’t analyze your intermediates for possible rearrangements, you can easily walk into a trap and lose points on the test by providing the wring answer.
Let’s look at a one more example:
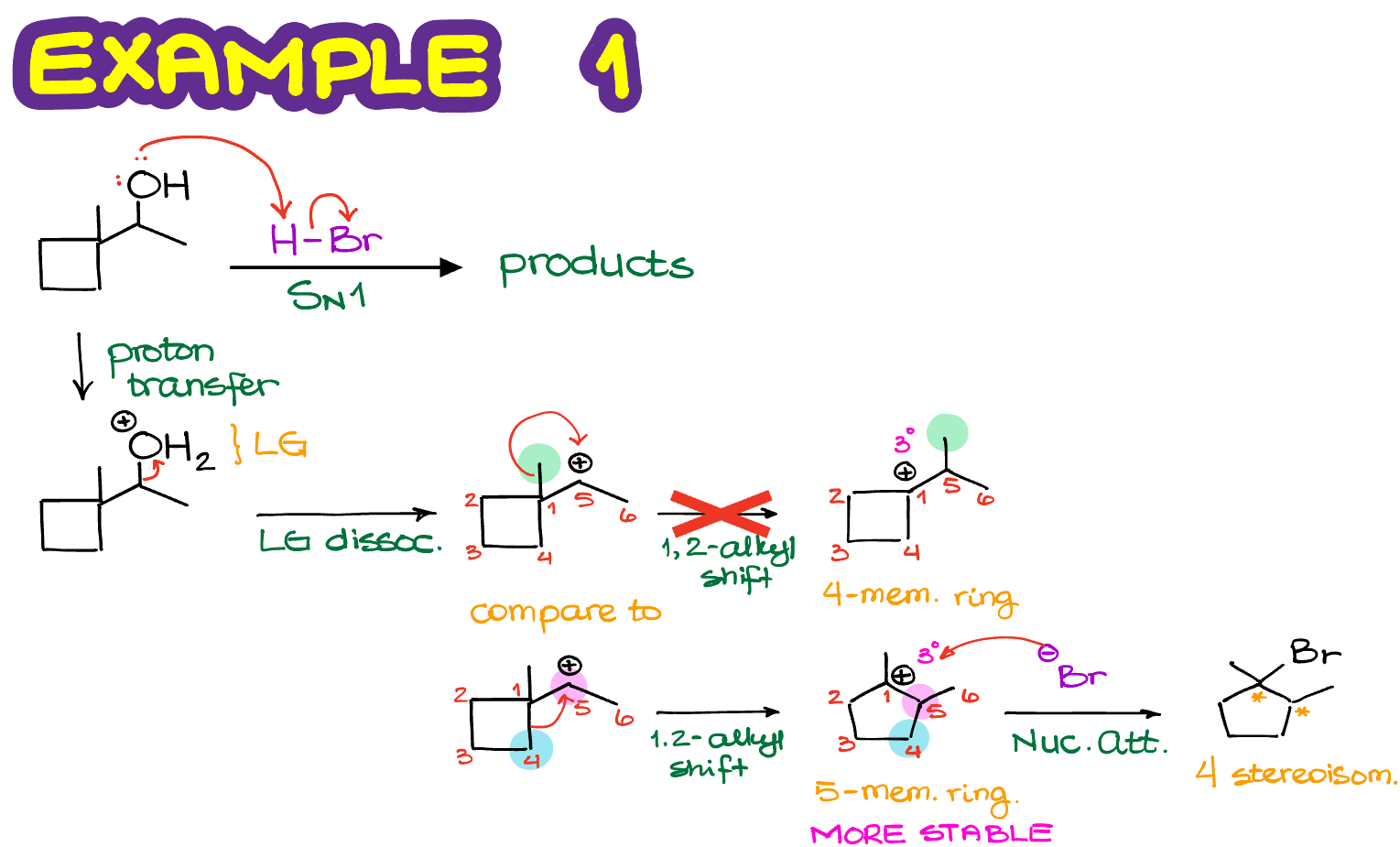
Here’s a reaction where we have a cyclic starting material with a good leaving group. Let’s first write the complete mechanism for this reaction.
In the first step, we are going to have our leaving group (H2O+) dissociate forming a corresponding carbocation. The carbocation that we form is secondary and it’s next to a quaternary atom. This means that we can have a carbocation rearrangement making a more stable carbocation intermediate. As we do not have any hydrogens on the 4° atom, it’ll have to be an alkyl shift. In the course of this shift, we can either move the methyl group onto the adjacent atom and make a new carbocation or go through the ring expansion. In this case, the ring expansion makes an overall more stable carbocation, so it’s going to be the major pathway for our reaction.
Next, we’ll proceed with the nucleophilic attack on our carbocation with the nucleophile that we have available to make the final product.
Notice, that in this case we are going to end up with two stereocenters. And as the SN1 reactions scramble stereochemistry of anything that they touch, we are going to end up with a total of 4 stereoisomers in this reaction. I challenge you to draw them all and assign the relationship between them. Assigning the R/S stereodescriptors first might be helpful. Hint: you should end up with two pairs of enantiomers and 6 pairs of diastereomers if you look at all possible combinations of the relationships.
Practice Questions
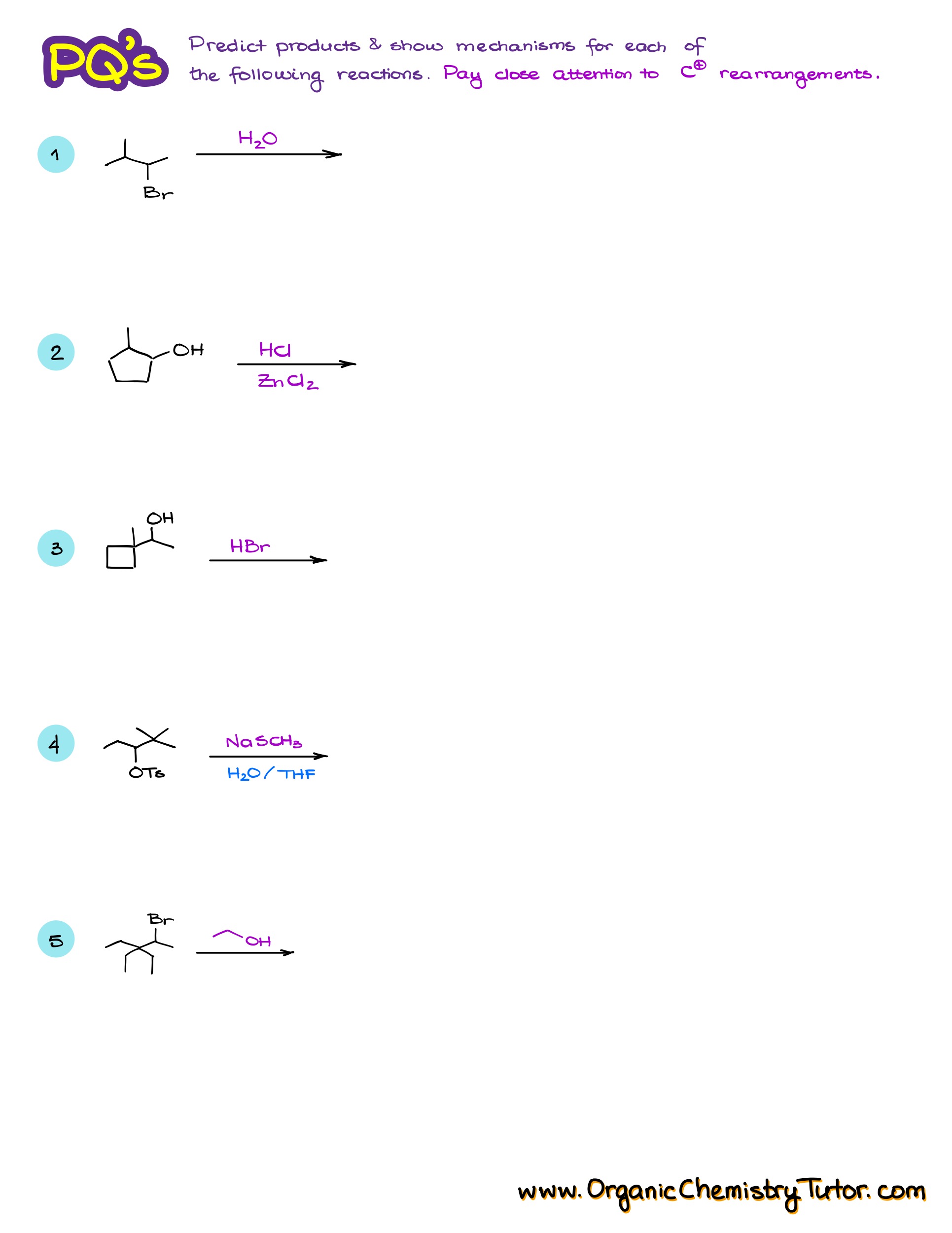
Hi! I think the answer key posted is for a different set of questions. Could you please look into this?
Thank you for letting me know. Fixed it.