Carbocations: Stability and Rearrangements
Welcome to today’s deep dive into the world of carbocations. By the end of this tutorial, you’ll grasp the intricacies of their structure, understand their stability, and unravel the mysteries of carbocation rearrangements.
What Are Carbocations?

At its core, a carbocation is an sp2-hybridized, 6-electron species sporting an empty p-orbital. Picture this: it’s like a jigsaw puzzle with a missing piece. This incomplete octet renders carbocations quite unstable. But there’s more – they’re also incredibly electrophilic, meaning they have a penchant for electrons. Why? Simply because they’re short of a full octet, making them electron-craving.
Stabilizing the Unstable
Now, while carbocations may sound like volatile rebels, nature has its ways of bringing stability. Two primary methods come to the rescue:
- Resonance with adjacent atoms’ electron pairs.
- Hyperconjugation with nearby sigma bonds.
A Quick Visual
Imagine a flat triangle, each corner representing an atom or a bond. That’s the trigonal planar geometry of a carbocation. And if you’re a fan of Star Wars, it somewhat resembles the imperial cruisers. A fun way to remember, right?
Carbocation Classification: From 1° to 3°
In the carbocation universe, they’re categorized based on their connections to other carbon-containing groups, aptly termed as R groups. We can classify them into three main categories: primary, secondary, and tertiary.
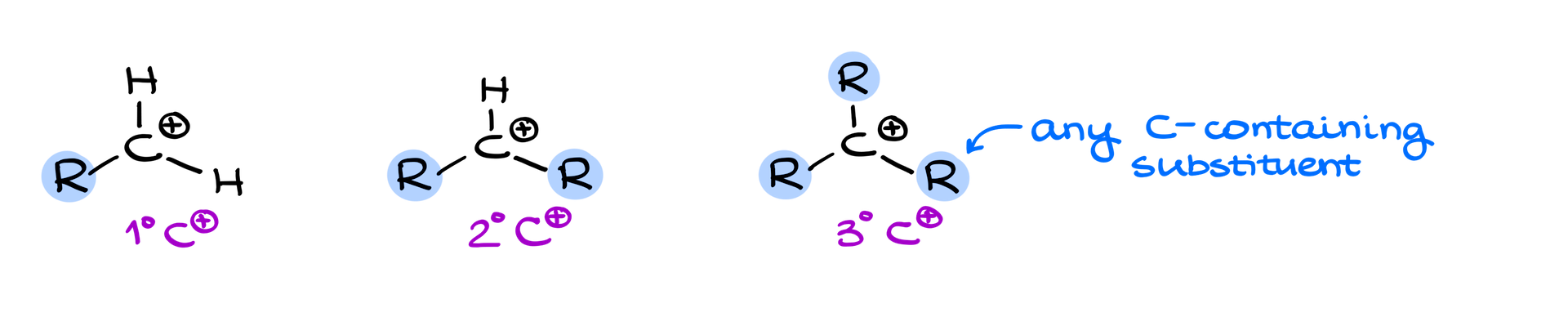
1° Carbocation (Primary)
Picture a carbocation linked to just one carbon-containing group. It could be something as simple as a methyl group or a more complex group. The key takeaway? Only one carbon connection.
2° Carbocation (Secondary)
These have two R group connections, typically leaving just one hydrogen behind. Think of it as the middle sibling in the carbocation family.
3° Carbocation (Tertiary)
The tertiary ones are like the elder sibling, linked to three R groups. These carbocations are surrounded by carbon-containing buddies on all sides!
Quaternary Carbocations: A Myth?
Now you might wonder, why isn’t there a quaternary carbocation? Imagine a carbocation connected to four different groups. Sound feasible? Take a moment, ponder, and share your thoughts in the comments below. Let’s get that discussion going!
The Pathways to Carbocations
Once you understand what carbocations are, the natural query is, “How are they produced in reactions?” Typically, two primary mechanisms lead to the formation of these charged species:
1. Leaving Group Dissociation:
Imagine a molecule with a leaving group like bromine (Br). Given the right circumstances, bromine can exit, leaving behind a carbocation.

But what instigates this exit? Bonds don’t just sever on a whim. If they did, we wouldn’t exist as structured beings! Bond dissociation needs a reason. Often, this reason is the presence of a polar protic solvent such as water, alcohols, or certain acids. These solvents stabilize the leaving group, ensuring it doesn’t make a U-turn and reattach to the carbocation.
If a polar protic solvent isn’t available, there’s a backup: strong Lewis acids. For instance, aluminum bromide (AlBr3) can complex with the leaving group, forming a new, more stable leaving group. When this super-charged leaving group departs, it leaves behind a carbocation ready to play its part in subsequent reactions.
2. Electrophilic Attack on a pi-Bond:
Alkenes, despite their ‘fragrant’ reputation, can be prime targets for hungry electrophiles like HX acids (e.g., HBr). During an attack, the pi-bond grabs a proton, resulting in a carbocation.
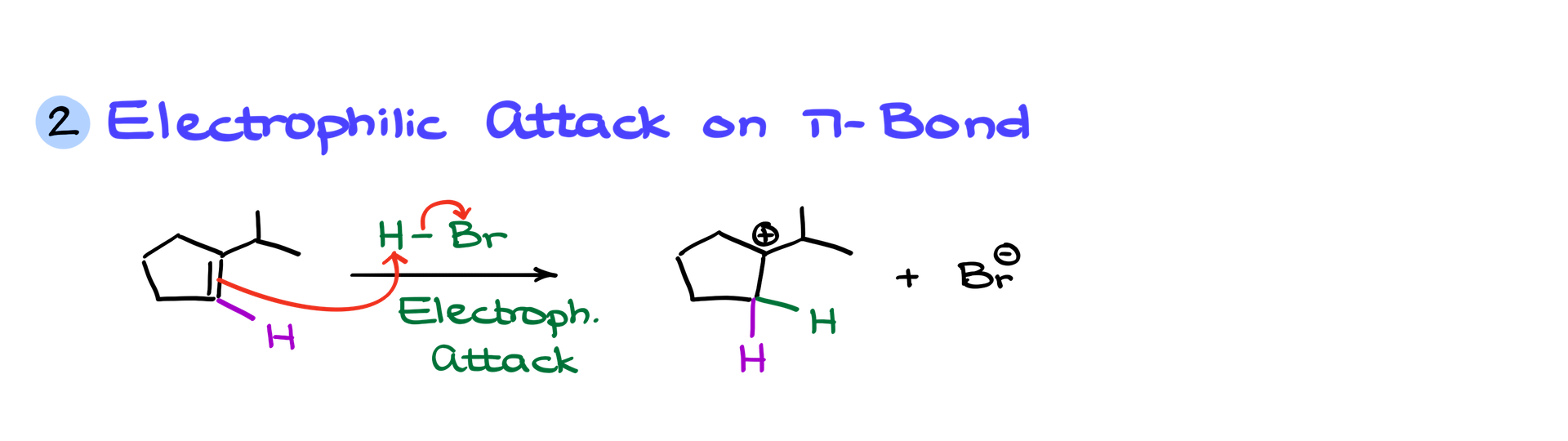
“But where did the hydrogen go?”, you might ponder. It’s still there but has now joined forces with another hydrogen atom. It’s like a reunion, where the proton from HBr finds an old buddy and together they lead to the formation of a tertiary carbocation.
Carbocation Stability
Imagine you’re at a picnic with a basket of apples. You reach out for an apple (which represents a carbocation), and you find that some apples are fresh and firm, while others are soft and squishy. Why is that? The environment around the apples and the factors affecting them (like the sun, rain, and air) are like the factors influencing carbocation stability.
Carbocation Stability: Not all apples are the same, and neither are all carbocations. Their stability varies, and two main factors play a role in it: resonance and hyperconjugation.
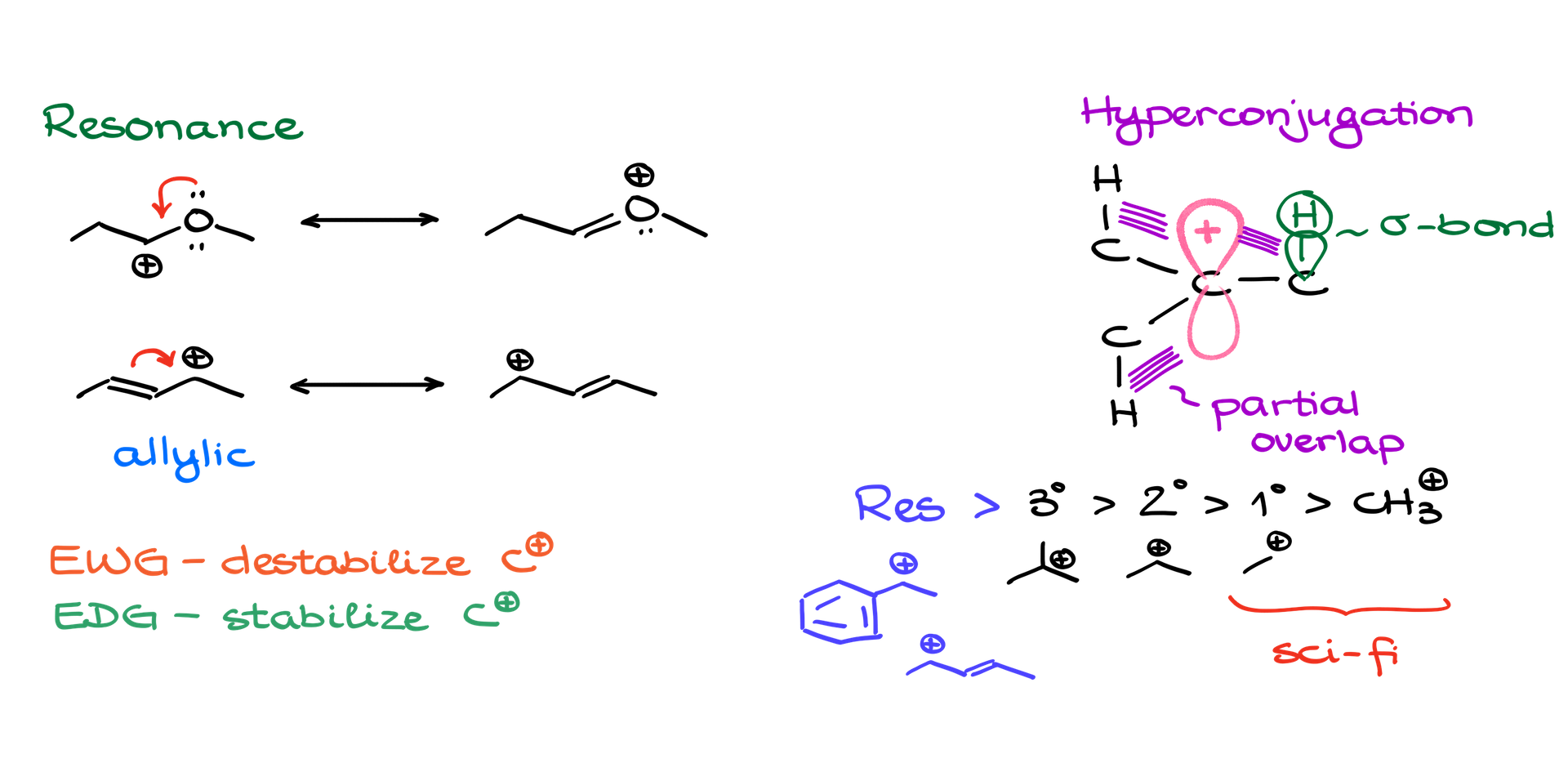
Resonance:
Imagine a tightrope walker (a carbocation) trying to maintain balance. If the walker has a net or safety ropes (representing electron-bearing elements), it adds stability. Carbocations are stabilized when they are adjacent to elements with electron pairs like oxygen, nitrogen, or sulfur. This is because these elements share their electron pairs, leading to a stable resonance structure. Another kind of resonance happens when a carbocation is next to a double bond, making it an allylic carbocation. Though not as strong as the first type, the more you have, the better. Think of it like multiple safety ropes for our tightrope walker!
Hyperconjugation:
Now, imagine you’re trying to light a campfire, and the wind keeps blowing out your match. You try to shield it with your hand or a piece of cardboard, but it only provides partial protection. This shielding effect is similar to hyperconjugation. When there’s an overlap between a sigma bond (like C-H) and the empty p orbital of a carbocation, it’s partial – like the cardboard shield. The more overlapping bonds you have, the more stable the carbocation becomes. So, a carbocation surrounded by many C-H bonds is more stable than one with fewer.
Hierarchy of Stability:
A quick real-life analogy: Think of carbocations like buildings. A skyscraper (tertiary carbocation) has a stronger foundation than a two-story building (secondary carbocation), which in turn is more stable than a shack (primary carbocation). And then there’s a tent (methyl carbocation) – that’s unstable and can easily fly away in the wind!
Remember our tightrope walker? Well, resonance (safety ropes) beats everything! Carbocations stabilized by resonance, like benzylic or allylic carbocations, are more stable than any other types, including tertiary carbocations.
Electron-Withdrawing vs Electron-Donating Groups:
Let’s switch gears and think of a carbocation like a hungry person craving electrons. If you put this hungry individual near someone hogging all the food (electron-withdrawing group), it will worsen their situation. But if they’re near a generous soul sharing their food (electron-donating group), they’ll feel better!
Carbocation Rearrangements: The Unexpected Turn in Synthesis Planning
Have you ever laid out plans and then something unexpected threw a wrench in the works? Well, carbocations are like that. Just when you think you’ve got them figured out, they shuffle around and leave you wondering what just happened.
I often talk about how unstable carbocations can be. But there’s another facet to their character that can be both fascinating and infuriating: the carbocation rearrangements. Imagine having your charged particle, the “plus,” hopping around your molecule, potentially skewing the outcome of your carefully planned synthesis.
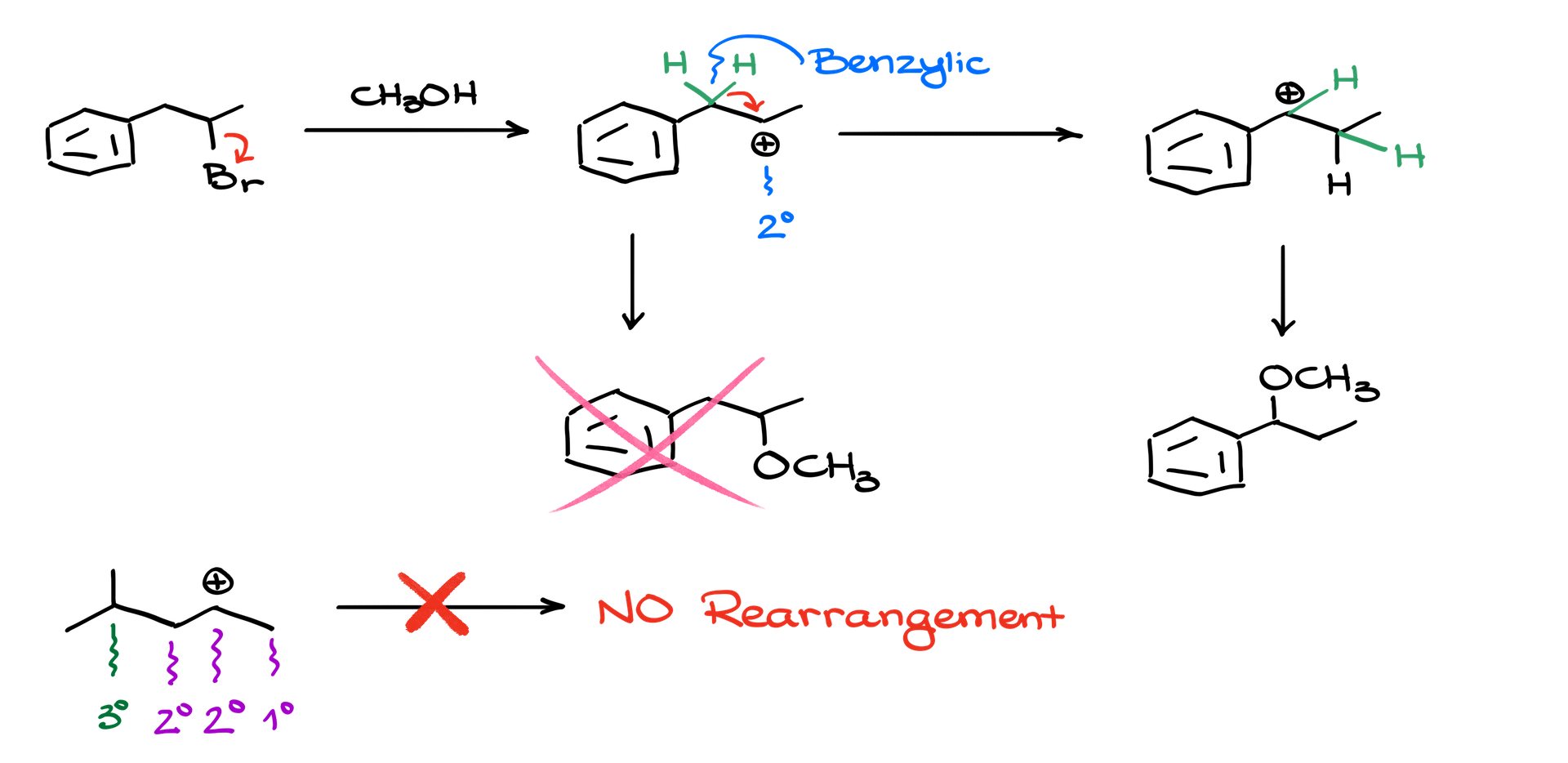
Let’s dive in with an example to better understand what I’m talking about. Picture a reaction where the first step involves a leaving group breaking away. This leaves behind a carbocation. It’s a secondary one, to be precise. But here’s the twist! Right next to this carbocation, there’s another secondary position that’s not just any regular one – it’s benzylic. Now, for those of you who’ve been on this chemistry journey for a while, you’d know that a benzylic position is way more stable than a regular secondary position. So, what does our carbocation do? It seizes the opportunity and rearranges itself!
Think of this rearrangement as a mini migration. A hydrogen, along with all its electrons, shifts over to the carbocation’s position, leading to the creation of a new carbocation. This fresh carbocation is right next to our aromatic ring and is stabilized by resonance, making it a significantly more favorable candidate for reactions.
In the realm of organic chemistry, this migration is termed the “1,2-hydride shift.” It’s as if we’re gently nudging the hydrogen to a new position. But here’s the kicker. This rearranged carbocation can result in an entirely different product than initially intended. To paint a clearer picture, if you were hoping for a particular ether as your product, the carbocation rearrangement could very well lead you to a completely different ether!
Let me leave you with a nugget of wisdom from the archives of organic chemistry. It’s estimated that carbocation rearrangements occur in about 90% of molecules before any other reactions kick in. So, the next time you’re strategizing a synthesis, always account for the potential twists and turns introduced by carbocation rearrangements.
Carbocations: Living in the Moment
Let’s pause and have a little fun analogy. Imagine if you went to the grocery store with only $10. You’d buy what you can right now, rather than thinking about a bigger purchase next month, right? Similarly, carbocations live entirely in the present moment. They don’t plot out long-term strategies, rather they act on immediate benefits.
Here’s the crux: A carbocation will opt to rearrange only if it can instantly achieve greater stability. If there are multiple rearrangements needed to reach the most stable state, well, the carbocation isn’t aware of that long game. It only responds to what’s immediately beneficial.
Picture this molecule in your mind. The carbocation here is secondary. To its immediate left and right are another secondary position and a primary position, respectively. But if you travel a carbon further, there’s a tertiary position beckoning. Tempting, isn’t it? Well, not for our carbocation. Our little charged entity is completely oblivious to this tertiary position. It’s like having a golden ticket just out of reach, but not knowing it exists. So in this scenario, there will be no rearrangement because our carbocation doesn’t “see” an immediate boost in stability.
In a Nutshell: Carbocations act on the here and now. They’ll rearrange only if they spot an instant boost in stability, blind to any long-term benefits. Keep this in mind as you navigate the labyrinth of organic reactions!
Alkyl Shift in Carbocation Rearrangement
Consider this molecule: it possesses a secondary carbocation next to both a quaternary and primary position. Primary carbocations are highly unstable and are rarely observed unless resonance stabilization is in play. Therefore, a carbocation at the primary position isn’t a feasible option. However, while a quaternary carbocation sounds appealing for increased stability, it’s physically impossible due to structural constraints.

So, what’s the solution? The molecule can undergo an alkyl shift. Instead of a hydrogen shift, the CH3 (methyl) group moves, transferring its electrons to the carbocation-bearing carbon. This shift transforms our structure. The original secondary carbocation becomes tertiary, offering a more stabilized structure. The quaternary position also transitions to a tertiary state. Through this alkyl shift, the carbocation successfully jumps from a secondary to a more stable tertiary position.
Group Migratory Aptitude in Carbocation Rearrangements
When it comes to carbocation rearrangements, not all groups have the same propensity to migrate. Group migratory aptitude is the tendency of specific groups to shift during a rearrangement. Among various groups, hydrogen is often the most inclined to migrate. So if a molecule has a hydrogen atom in proximity to the carbocation, it will likely shift first.
Next in line is the phenyl group, which is essentially an aromatic ring. After the phenyl group, the migratory aptitude generally follows this order: tertiary alkyl groups, secondary alkyl groups, and then primary alkyl groups.
As a general guideline, the order is: hydrogen > phenyl group > alkyl groups (tertiary > secondary > primary). In practical applications, especially in classroom settings, it’s rare to face a situation where the choice is between a tertiary and a primary alkyl group. It’s because the actual outcome might necessitate experimental data to determine.
Carbocation Rearrangement with Resonance Stabilization
Consider a molecule with a carbocation at a secondary position. At first glance, the surrounding carbons—both secondary—might suggest no possible improvement in stability. However, it’s essential to note that even if electron pairs aren’t always explicitly depicted, they’re present, such as on an oxygen atom.
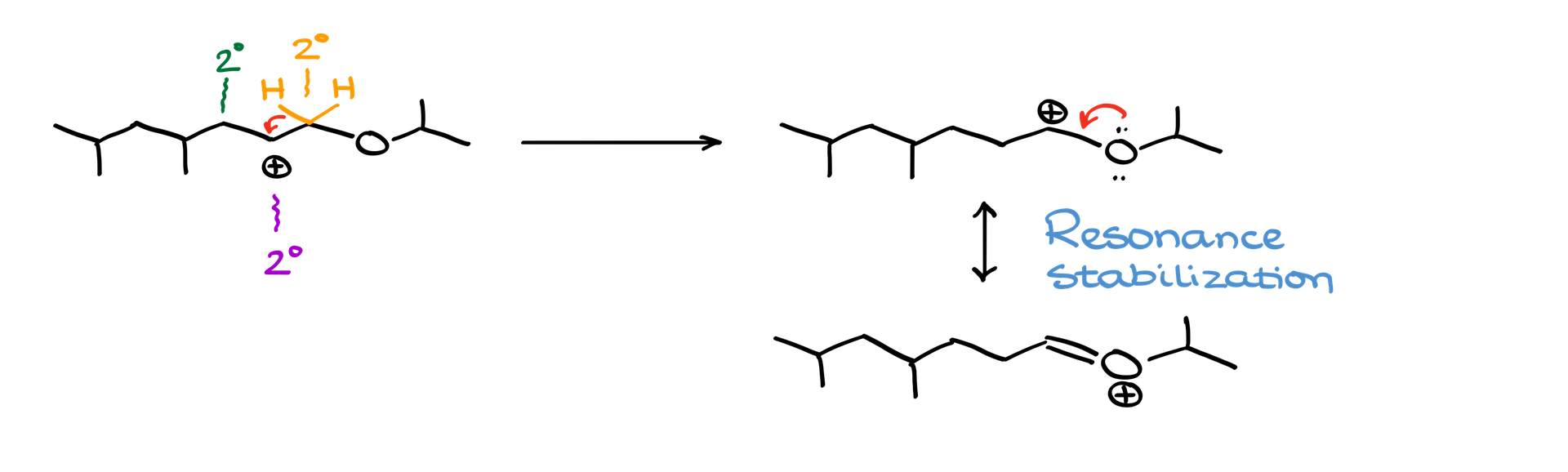
In this example, if the carbocation is adjacent to an oxygen atom, the oxygen can stabilize the carbocation via resonance, providing a motive for rearrangement. To achieve this, one hydrogen, along with its electrons, shifts towards the carbocation, forming a new carbocation. The new position now gets stabilized by the oxygen’s lone pair via resonance.
Drawing all implicit hydrogens and electron pairs can aid in visualizing such rearrangements more clearly. In the given rearrangement, the oxygen’s lone pair stabilizes the new carbocation, resulting in a positive charge on the oxygen. Although this might seem concerning, both the oxygen and carbon have complete octets, rendering the molecule stable. This configuration is the major resonance contributor, making it more stable than the original carbocation.
It’s crucial to note that only nearby positions influence the carbocation. In this scenario, a distant tertiary position doesn’t impact the carbocation’s behavior, making it effectively non-existent in the rearrangement context.
Concluding Thoughts
So, to recap whenever you form a carbocation in your reaction always check for possible rearrangements. They are always sneaky and they’re always going to mess up your synthesis if you don’t remember about them. Typically over 90% of molecules will rearrange before they react with a nucleophile. Once you’ve checked your carbocation for any rearrangements, did those rearrangements, and your carbocation as stable as it can be given the molecular structure of your compound, then you can proceed your reaction.
Sir, here at the start you have mentioned that carbocation is sp² Hybridized but I’m wondered about CH5+(methanium), what would be its Hybridization?
That species doesn’t fit into a typical valence bond model as it exhibits non-classical bonding patterns. It’s a highly unstable species and there’s no empirical data on its actual structure outside of two theoretically postulated possible bonding patterns for it including a 3-center-2-electron bond similar-ish to what we see in boranes. And since hybridization is a mathematical model that’s only applicable towards the classic 2dn period bonding patters, it’s pointless to even try to come up with a reasonable hybridization for methanium ion.