Halogenation of Ketones and Haloform Reaction
In this tutorial I want to talk about the ⍺-halogenation of ketones and the haloform reaction. But before we go into the discussion of the reaction itself and see how that happens in both basic and acidic conditions, let’s take another look at enols and enolates first.

In the figure above, we have the resonance structures for the enol. On the left, we have the major contributor, and on the right side we have the minor contributor. The left structure is the major contributor because we have no charges on it. The minor contributor, however, does have charges on both oxygen and the carbon. By putting both of those two contributors together and sort of like averaging them out in a hybrid form, we would notice that the carbon atom will have a partial negative charge which makes that carbon nucleophilic.
Now let’s compare those two resonance structures to the similar resonance that we have in an enolate species.

The very first thing that we notice right away about the enolate species is that it has a negative charge. This means that we have the excess of the electron density on the enolate species. Thus, enolates are significantly more nucleophilic than enols. And since they’re more nucleophilic, that means that they’re going to be more reactive in reactions with electrophiles. Another thing that we notice here is that we have two places in the molecule where the negative charge is going to be. One place is on the oxygen and another one is on the carbon. That means that an enolate species actually has two nucleophilic regions in the and there are two possible reactions that we can have with an electrophile. Typically, the carbon atom plays the role of a nucleophile in reactions of enolates. However, occasionally it is possible to have oxygen playing the role of a nucleophile. Those reactions are a little bit more rare but we will discuss specific factors that will influence carbon versus oxygen nucleophilicity in one of our future tutorials.
Halogenation in Acidic Media
Now, when we have reminded ourselves how enols and enolates look like and which part of the molecule is nucleophilic, let’s look at some of the reactions of enols and enolates. The first reaction that I want to look at is going to be the halogenation in acidic media. Here is the general scheme for the acid-promoted halogenation reaction we are going to be reacting our carbonyl compound ketone acetophenone in this case with the halogen reaction.
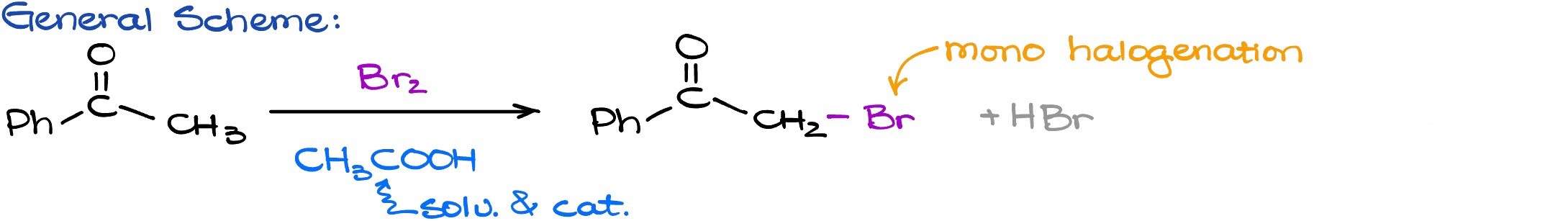
For this example, I am going to be using the Br2. We also have acetic acid here as the acidic catalyst that is not going to be consumed during our reaction. For the product, we have the ⍺-position where the one of the hydrogens has been substituted with the bromine atom. We’re also going to be making HBr as a co-product which we don’t really care about.
Let’s look at the mechanism of this reaction.
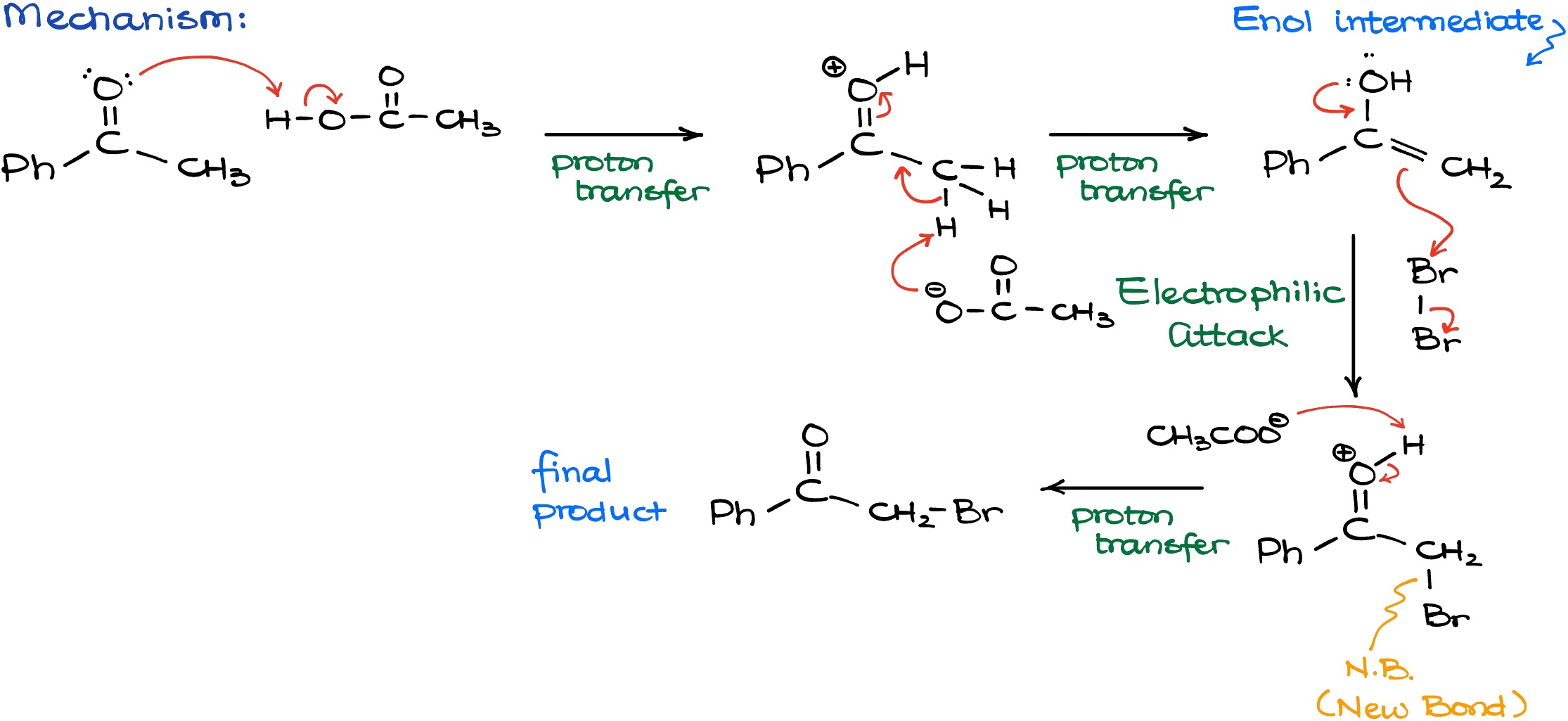
The very first step is going to be the protonation of our carbonyl compound with our acidic catalyst. We need this step to convert our ketone into a corresponding enol species. And once I have my protonated carbonyl, I can use the conjugate base from the acetic acid to deprotonate it and finally convert it into my enol. Once I do have my nucleophilic enol intermediate, I can proceed with the reaction with the halogen, Br2. The reaction with Br2 going to create a new carbon-bromine bond and break a bromine-bromine bond. I’ll show that on the scheme above as the “N.B.” abbreviation. As the result of this interaction, we’ll have a protonated intermediate that is not particularly stable. So, to get our final product, we are going to deprotonate this intermediate by using some sort of a conjugate base from the solution. For instance, I can use my Br– or anything else with an electron pair floating around. That would yield me my final product.
Kinetics of the Acid-Promoted Halogenation
When it comes to the reaction kinetics we know (from the experimental evidence) that the rate law in this reaction only depends on the concentration of the ketone and the concentration of the acid. Thus, this is a second-order process. Which means that the reaction is independent from the concentration of the halogen. So if I rewrite the mechanism of my reaction here, I’ll get the following scheme.
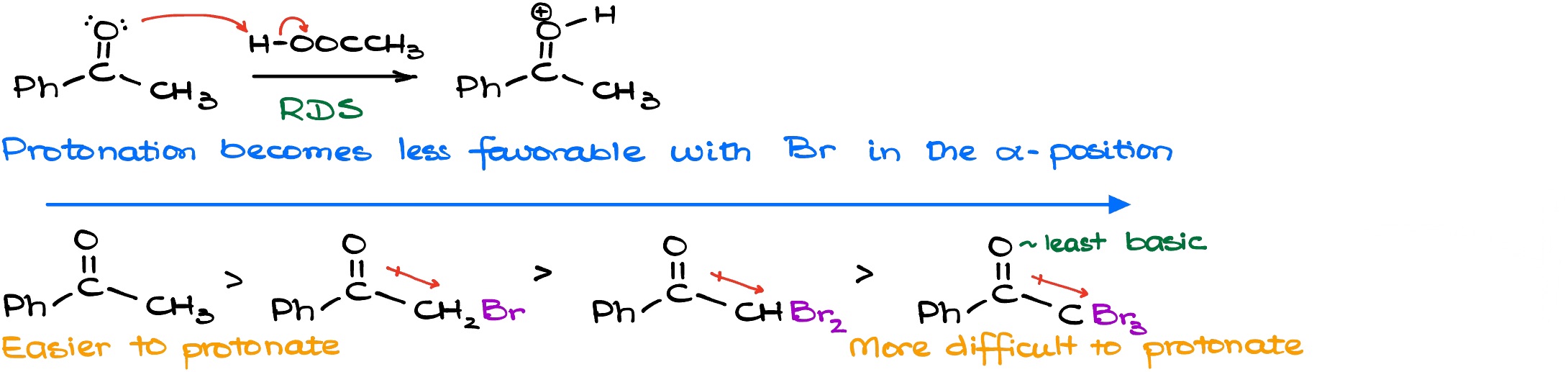
Based on the rate law and the experimental evidence, we can say that our rate determining step is the enolization process itself. So either the step number one or the step number can be determined as the rate determining step (RDS or rate limiting step, RLS). Based on the experimental evidence we know that’s typically going to be the first step. Thus, the protonation itself is the slowest step on this reaction. Here’s something that’s very important: because as we are adding bromines to our molecule the molecule becomes less and less basic. So, the resulting carbocation species that we get after the protonation becomes less and less stable the more electron withdrawing groups we have. In other words, the species become less stable the more halogens we have. Halogens are relatively electronegative species and is pulling the electron density towards itself via the induction. So, if I have my bromine pulling electron density towards itself, the electron pair on the oxygen atom is going to be significantly less nucleophilic/basic. This way, as we add more halogens to the molecule, the reaction becomes slower, and slower, and slower with every extra halogen. And for the purposes of the everyday chemistry, we can say that the reaction essentially stops after the addition of the first halogen.
Base-Promoted Halogenation
If we look at the base catalyzed reaction, on the surface it looks extremely similar to acid catalyzed reaction.
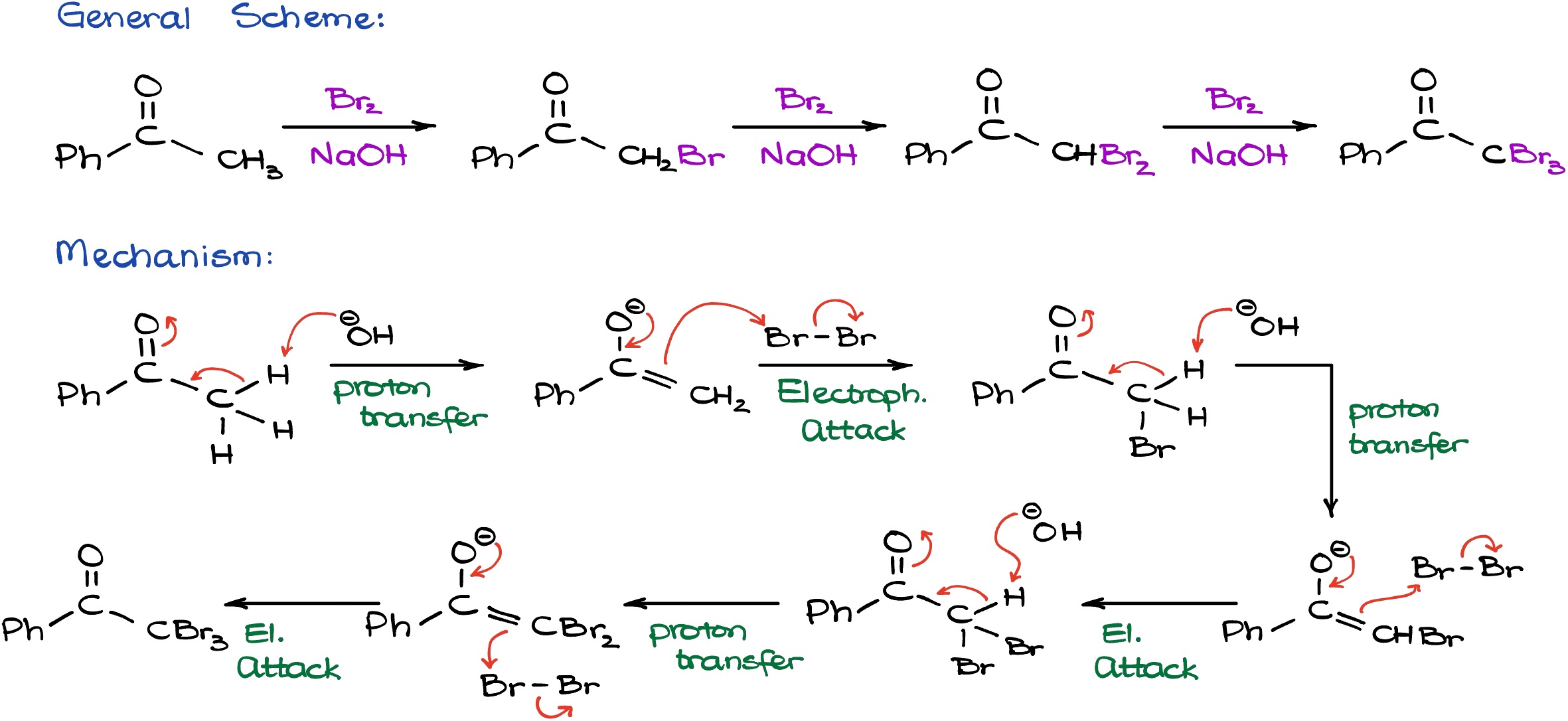
However, as a catalyst we are going to be using sodium hydroxide. And unlike in the previous case, sodium hydroxide in reality is not a catalyst but a reagent in this case because it is actually going to be consumed and it’s never going to be regenerated as a part of this reaction. The mechanism for this reaction will start by the formation of the enolate species. Since sodium ion is a spectator ion, we don’t really care about that. In the first step, the OH- is going to deprotonate our molecule giving us an enolate species looking. We know that enolates are extremely nucleophilic, so when we have an electrophile around we’re going to see the reaction between them. In this reaction, the carbon atom and the bromine atom are making a new carbon-bromine bond similar to how we did this reaction in the acidic conditions. However, here we are actually going to form the final product right the way and we’re going to have the Br- as a co-product which is going to recombine with sodium giving us sodium bromide as the co-product (which we don’t really care about either).
Here is something interesting about this reaction though: as soon as I add Br to my molecule, the protons at the alpha position of my carbonyl actually become more acidic than what they were be at the beginning of my reaction. This means that if we have an excess of the base, the reaction is going to continue and the second round of deprotonation is going to be even easier. And likewise, once we form the enolate species, for as long as we have the electrophile around, we can easily continue this reaction and make the second carbon bromine bond. Now, as soon as we add the second bromine to the molecule, due to the inductive effect, the leftover protons are going to be even more acidic than in the predecessor. In this particular case we only have one proton left, but you get the idea. This means that the reaction is going to proceed even more favorable and even faster for the last deprotonation and formation of the new enolate species. Which, in turn, is going to proceed making the third bromine-carbon bond resulting in a product where every single ⍺-proton is actually substituted with a halogen. In other words, we are going to have an exhaustive halogenation of our molecule. So, whenever you are trying to do the base promoted halogenation you cannot stop at the information of the monohalogenated product and the reaction is going to keep going. If you try to do your reaction in a one-to-one ratio in the conditions where the Br2 and sodium hydroxide or any other hydroxide that you might be using are not used in the excess, you’re just going to get a mixture of all possible products. So, these reactions are typically done in an excess of the halogen and excess of a base. And all hydrogens that we have in the ⍺-position are going to be substituted with bromines.
The Haloform Reaction
If you end up with the CX3 group, where X is a halogen, the reaction doesn’t stop there either. This is known as a haloform reaction. The CX3 is a potential living group. And if you have a leaving group on a carbonyl, you can easily do an acyl substitution. In addition, the trihalogenated group is a strong electron-withdrawing entity and will make the carbon atom of a carbonyl extremely electrophilic.
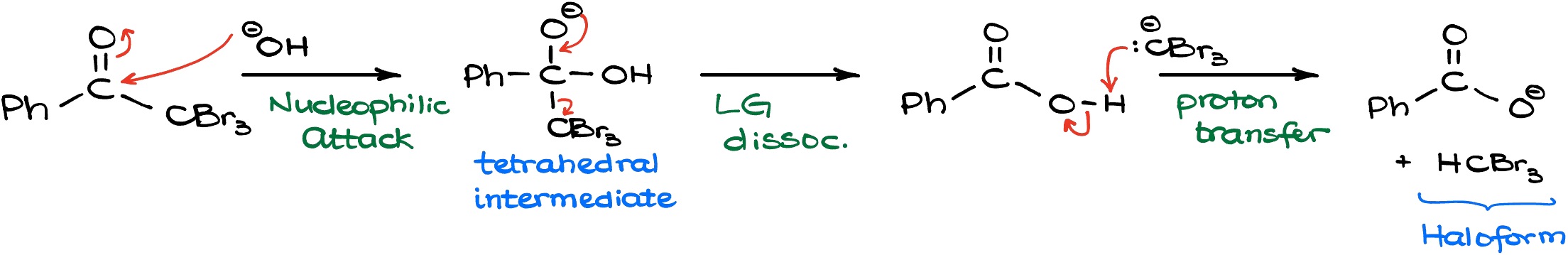
The attack of the hydroxyl on the carbonyl gives us a tetrahedral intermediate which is very common for carboxylic acid derivatives. Most of their reactions happen through the formation of some sort of tetrahedral intermediate. The tetrahedral intermediate, in turn, is going to displace the living group which is the CBr3 in this case. And as that happens, we’re going to form a carboxylic acid and a Br3C- species which is fairly basic. So, since they form right next to each other the Br3C- will instantly snatch off the proton of our carboxylic acid and deprotonate it. In this final proton transfer we get the corresponding carboxylate and HCBr3 which is referred to as a bromoform. Any molecule with the generic formula of HCX3 is called a haloform which is where the name for this reaction comes from. Thus, if we have chlorine that’s going to be chloroform, if we have iodine it’s going to be iodoform, and bromoform for the version with bromine.
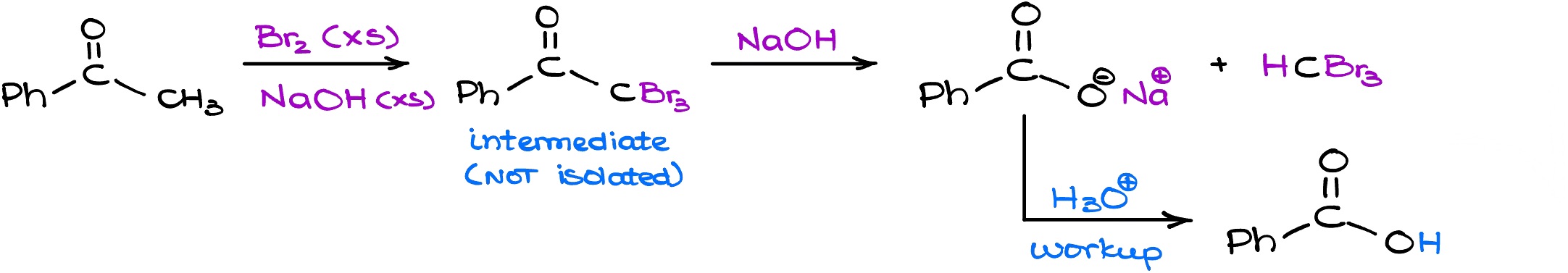
So if we look at the complete scheme for the haloform reaction, we see is that if we start with a carbonyl compound with a ketone with a methyl group on it and reacted with the bromine (or any other halogen in basic media) this reaction will keep on going until you end up with a carboxylate and the haloform. Typically, we’re going to follow up with the acidic workup and get the carboxylic acid as the product in this reaction.
Examples of ⍺-Halogenation and the Haloform Reaction
Let’s look at a few examples. Here I have the same starting material reacting with bromine in both acidic conditions (on top) and in basic conditions (on the bottom of the scheme).
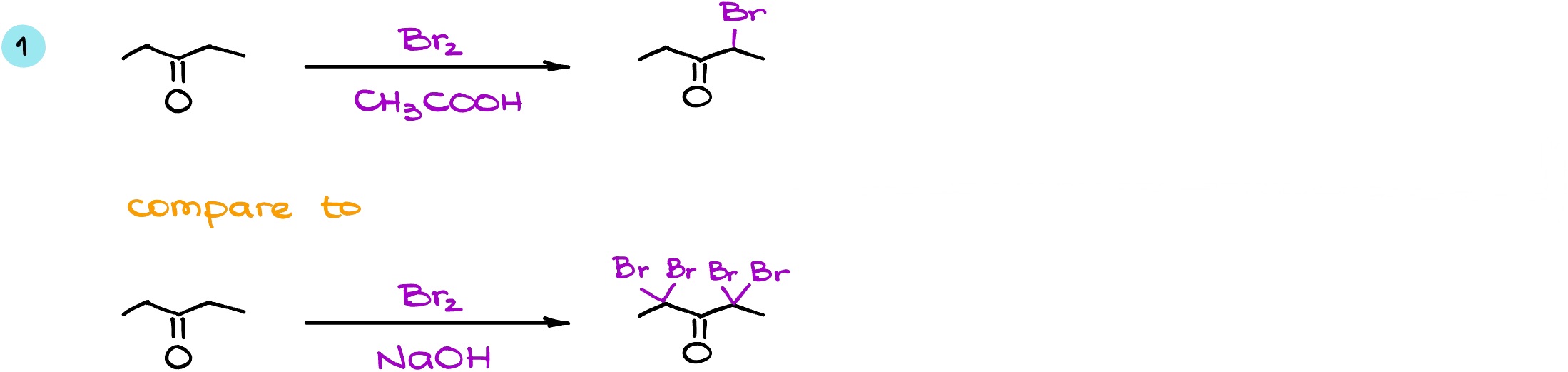
In the first case, I’m going to end up with only one bromine in the enolizable alpha position. As both enolizable positions are identical, it doesn’t matter which one we choose for the reaction. So in this case I’m going to get a monohalogenated product. In the basic conditions however, while I do have the same starting material with the same enolizable ⍺-positions the reaction goes to exhaustive halogenation. Thus, it does not stop after a single halogenation and in this case, we’re going to turn all of the hydrogens that we have in the ⍺-positions into the bromine atoms. And since we have CH2 groups in the ⍺-positions and not CH3’s, reaction does not continue as a haloform reaction. Instead, it stops at the formation of the tetrahalogenated product.
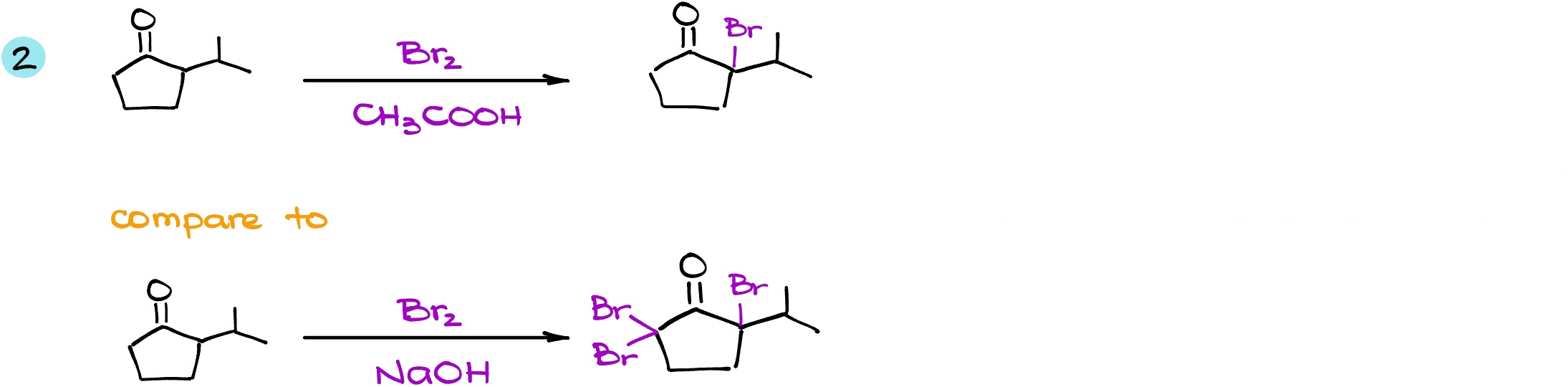
Here is another example. In this example, we are again going to contrast the same starting material but in different conditions: one in the acidic conditions, and another one in basic conditions. In the first case, we have two ⍺-positions, but now my molecule is no longer symmetrical. This means that we have one alpha position on the right (let’s call it a pink ⍺-position) and on the left (the green ⍺-position). In acidic conditions, we know that the enolization preferably gives us the more substituted enol. This means that my intermediate is going to be coming from the pink ⍺-position. When I do the same reaction in the basic conditions, while we still have the same pink ⍺-position and we still have the same green ⍺-position, in this case it doesn’t matter where we’re going to form the enolate. The basic conditions reaction will always give us the exhaustive halogenation, so the nature of the enolate is majorly irrelevant.
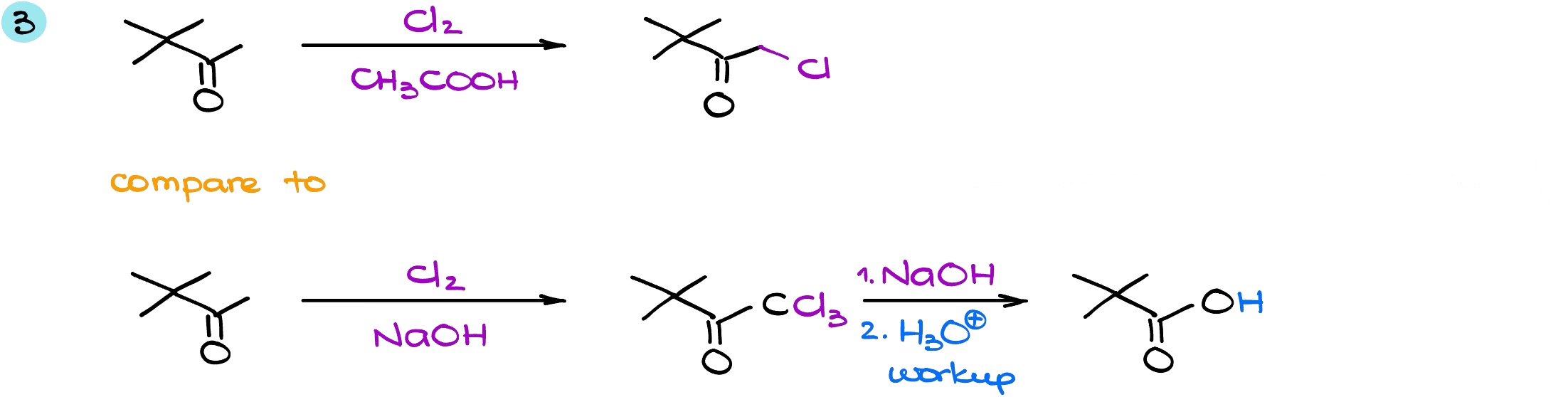
And in the last example, again, we have two of the same starting material in different conditions. While this reaction is with chlorine and not with the bromine, the idea is exactly the same. When I’m analyzing my molecule, I can see that in this case we do have the ⍺-position on the right (this pink ⍺-position) which is an enolizable position. However, on the other side (let’s say this is a blue ⍺-position) that is a non-enolizable position. And because of that, we’re not going to be considering that position for the reaction whatsoever: no protons there = non-enolizable position = we cannot form either enol or enolate. This means that position is pretty much non-existent for us in this reaction.
In the acidic conditions the reaction will only happen once. And once that happens, we add a single halogen to our molecule. Once we put our first halogen onto the molecule, that’s where it’s going to stop. In the second case, we are going to have the exhaustive halogenation which is going to give us CCl3 group. Since, it’s a leaving group, the reaction will continue to form a haloform and carboxylic acid.