Aldehydes and Ketones
Aldehydes and ketones are the two functional groups that share a lot of similarities. They both contain the C=O double bond, they both are polarized and have a δ+ charge on carbon and a δ- charge on oxygen. Thus, due to the structural similarity, aldehydes and ketones have many reactions that are the same for the both functional groups. That is why we often combine them in textbooks and lectures. So, what are the typical must-know reactions of aldehydes and ketones you want to know to ace your course? Here they are!
Reduction Reactions
There are several different ways how you can reduce and aldehyde or a ketone to a corresponding alcohol. While the reagents may differ, the idea is always the same: aldehydes reduce to primary alcohols, while ketones reduce to the secondary alcohols.
Reduction of Aldehydes and Ketones on a Heterogeneous Catalyst
Remember your reduction of alkenes or alkynes with H2 on Pd or Pt catalysts? Well, it’s kind of the same deal here.
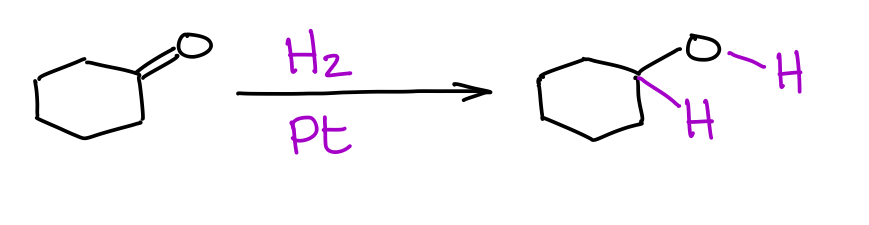
The main difference is that reduction of aldehydes or ketones generally requires a much higher pressure of hydrogen gas. Thus, you could reduce a double bond of an alkene without touching the double bond of a ketone or an aldehyde. Different compounds may not be as sensitive to the pressure of the hydrogen gas, so you always want to double check with your instructor how they would treat this reaction and its selectivity towards the C=O vs C=C bonds.
Reduction of Aldehydes and Ketones with Complex Metal Hydrides
Complex hydrides like lithium aluminum hydride or sodium borohydride are a perfect choice for the reduction of polar π-bonds like C=O.
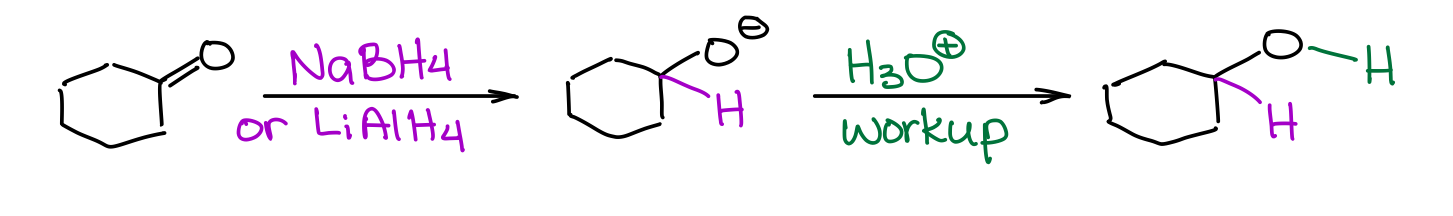
You must be careful though, as NaBH4 and LiAlH4 are quite different in their strength, so they are not 100% interchangeable. While NaBH4 is a mild reducing agent and can tolerate many other functional groups like carboxylic acids, esters, amides, or nitriles, LiAlH4 is an extraordinarily strong reducing agent and will react with almost any polar double or triple bond like C=O, C=N, or C≡N bonds. You’ll typically have an acidic workup with these reductions to protonate the resulting alcoholate species.
Wolff-Kishner Reduction
Wolff-Kishner is oldie but a goodie. This is a two-step process where you’ll make an intermediate imine compound which then goes through a cascade of acid-base steps, eventually yielding a completely reduced product.
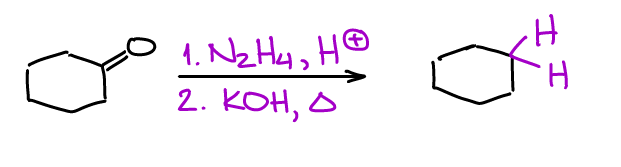
There’s also a one-pot version of this reactions which many instructors and textbooks tend to emphasize in their course. The major disadvantage of the Wolff-Kishner reaction is the temperature requirement. This reaction is typically done at about 250℃ which is too harsh for many organic substances. This also means that this reaction is not suitable for most light-boiling substances either as they will simply evaporate.
Clemmensen Reduction
A good alternative to the Wolff-Kishner reduction is the Clemmensen reduction. Clemmensen is done in much milder conditions and typically requires zinc amalgam and an acid (usually HCl or acetic acid).
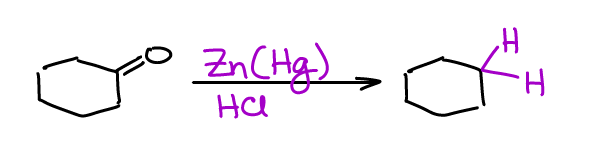
You could also use zinc shavings instead of an amalgam. However, the amalgam is much more reactive and generally yields better results. There are versions with other metals like iron (Fe) or tin (Sn). The zinc version is the most popular though.
Reduction via Thioacetal Intermediate
The last method is a reduction of thioacetals.
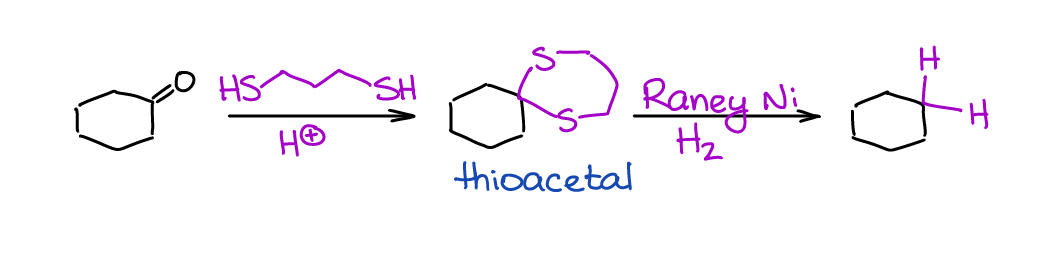
This is more of a synthetic sequence rather than a single reaction, but it’s often done in this sequence. Reaction is extremely selective towards aldehydes and ketones since you won’t be able to make a thioacetal with other C=O containing species. Thus, it can tolerate many other functional groups in your molecule. You also must use the Raney Ni and not your typical Pt or Pd catalyst. Raney Ni has a remarkably high affinity towards the S-containing substances and “rips” the sulfur off your molecule replacing it with H’s.
The disadvantages of this sequence are: (1) thiols are incredibly stinky 🤮, and (2) the catalyst will be essentially destroyed in this reaction and you won’t be able to reuse it in the future (it’s very cheap, so not a big deal).
Reactions with Organometallic Compounds
Aldehydes and ketones are excellent electrophiles due to the high δ+ charge on the carbon atom of C=O group. Thus, they react readily with many nucleophiles. Most common organometallic compounds are extremely nucleophilic, which makes them perfect candidates for this reaction. In a typical organic chemistry course, we go over the reactions of aldehydes and ketones with organomagnesium compounds (Grignard reagent) and organolithium compounds.
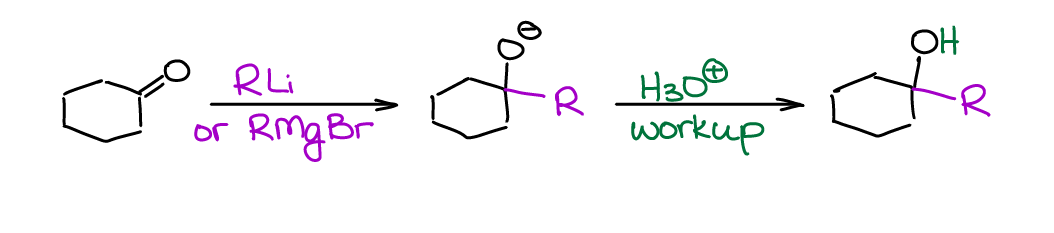
While there’s some difference in reactivity between the RMgX and RLi, we’ll treat them as pretty much the same type of reagent within the scope of a typical sophomore course. The Grignard reagent (RMgX) is a more common choice for these reactions though. So, you’re going to see it most often.
Reversible Nucleophilic Addition to Aldehydes and Ketones
Most nucleophilic addition reactions to aldehydes and ketones with O- or N-nucleophiles are reversible reactions. Depending on the nature of your nucleophile, you can get half-a-dozen functional groups, most of which show up in synthesis or biochemical processes.
Hydration of Aldehydes and Ketones
Hydration, or reaction with water, is the first example of the reversible equilibrium reaction of aldehydes and ketones you’re going to see in your course. In this reaction, the carbon of the C=O bond (an electrophile) reacts with water (a nucleophile).
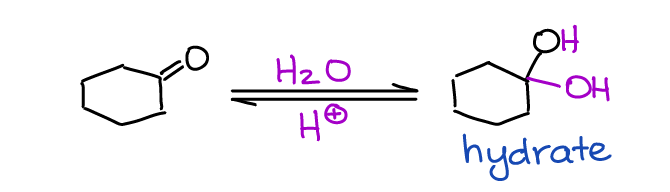
What’s interesting about this reaction, is that it can be catalyzed by both acidic and basic conditions. The hydrate is generally unstable. Thus, the equilibrium is shifted towards the starting material (carbonyl). There are several factors that can make the formation of the hydrate more favorable. Notably, electron-withdrawing groups tend to make hydrate more favorable by destabilizing the carbonyl itself.
Formation of Hemiacetals and Acetals
Hemiacetals and acetals are extremely important functional groups in organic chemistry and biochemistry. You’ll see them when we discuss the chemistry of carbohydrates and we’ll have a heavy use of acetals as protecting groups in multistep synthesis.
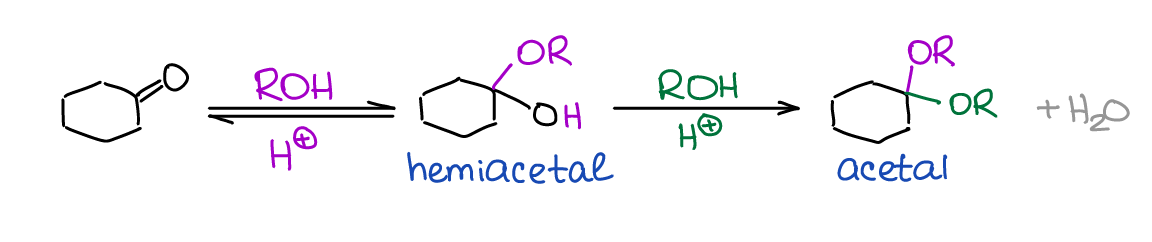
One important feature of hemiacetals is that they are generally unstable. While there are a few exceptions to this general rule, you’ll virtually never see a hemiacetal as a final product in a reaction.
Formation of Thioacetals
Thioacetals are the sulfur-containing analogs of the acetals.
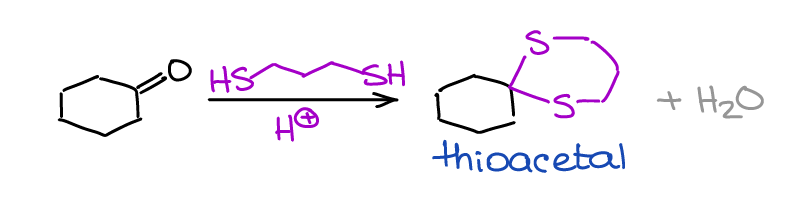
Thioacetals have a few limited uses in organic chemistry and synthesis. Within a scope of a typical sophomore organic chemistry course you’ll only encounter them once in a reduction reaction that I’ve mentioned earlier.
Formation of Imines
Imines are important intermediates in both biochemical processes and in organic synthesis.
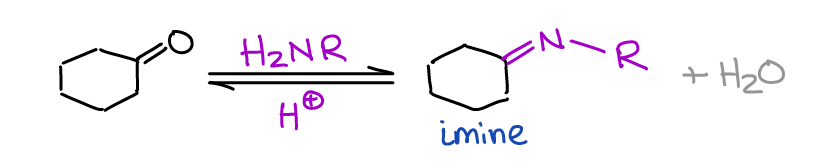
Very rarely we’re going to seek the formation of an imine as a final product, but as intermediates, they are invaluable! You make an imine by a reaction of a primary amine with an aldehyde or a ketone.
Formation of Enamines
Unlike imines, enamines are a big deal in organic chemistry and synthesis. You’ll see them later in your course as important intermediates in the reductive amination reactions and in the Stork enamine synthesis.
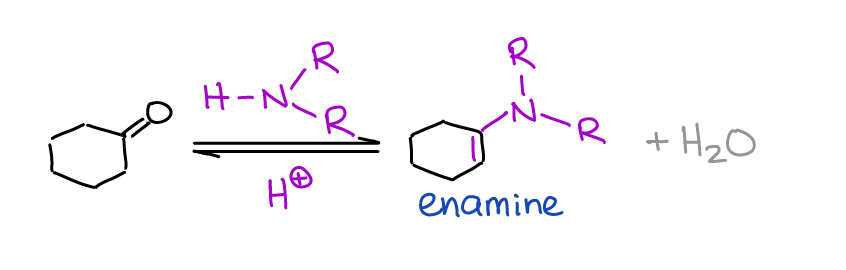
Enamines are formed by the reaction of the secondary amine with an aldehyde or a ketone.
Formation of Cyanohydrins
Cyanohydrins are a type of a functional group where you have the nitrile (-CN) and hydroxyl (-OH) on the same carbon in a molecule.
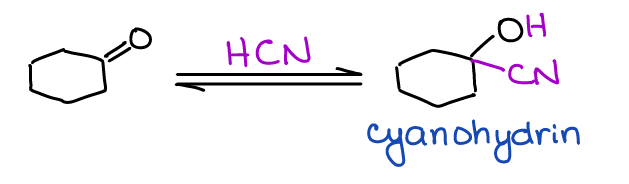
While cyanohydrins are not overly exciting, they do offer a neat route to the synthesis of carboxylic acids. One of the classic amino acid syntheses called Strecker amino acid synthesis uses a similar reaction.
Oxidation Reactions
While aldehydes and ketones do have the carbon atom in a rather high oxidation state, they can be oxidized further. There are two reactions that you’re likely to see in your course.
Baeyer-Villiger Oxidation
Baeyer-Villiger oxidation is a straightforward way to of converting a ketone into an ester.
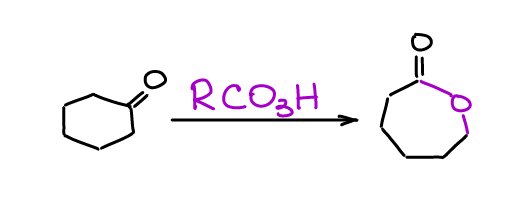
There are a few interesting aspects of this reaction that commonly show up on the test. First, the more substituted alkyl chain tends to migrate before the other alkyl chain in a ketone. Secondly, the stereochemistry of the migrating atoms does not change! While this reaction is most useful for ketones (and it sees a lot of industrial applications), you can perform it for aldehydes as well.
We use peroxyacids as the oxidizing agent in the Baeyer-Villiger reaction. The most common choice is mCPBA, however, other acids show up from time to time as well. The nature of the peroxyacid is, essentially, irrelevant here.
Oxidation of Aldehydes to Carboxylic Acids
When we deal with the oxidation reactions, we often see aldehydes as the midpoint products in the oxidation of the primary alcohols with Cr(VI) reagents (aka, the Jones Oxidation). There are, however, selective methods that can oxidize aldehydes to carboxylic acids without touching alcohols.

The most notable selective oxidizing agents for aldehydes are the Tollens’s reagent (AgO in aqueous ammonia) and the Benedict’s reagent (copper sulfate with a few other components).
Witting Olefination
Witting olefination or Witting reaction is a method of making the C=C bond in a single step from an aldehyde or a ketone and a Wittig ylide. This reaction is so powerful and versatile, that it got Georg Wittig a Nobel Prize in 1979.
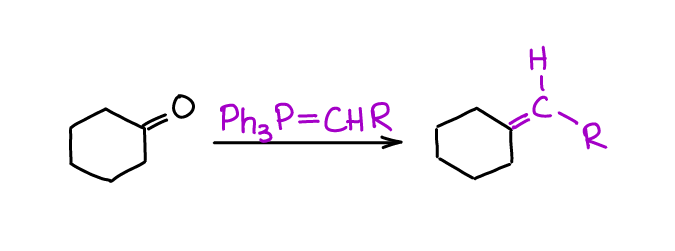
Since Wittig reaction is so powerful and tolerant of other functional groups, you’re going to see it on your homework and in the multistep synthesis questions in your course.