Alkenes
Reactions of alkenes is a huge topics. Alkenes, as a functional group, is a very versatile one. You can reduce it, you can oxidize it, you can cleave it, and you can do a large number of various addition reactions modifying an alkene to other functional groups.
So, let’s go over the must-know reactions of alkenes that you want to know to ace your next exam!
Catalytic Hydrogenation of Alkenes
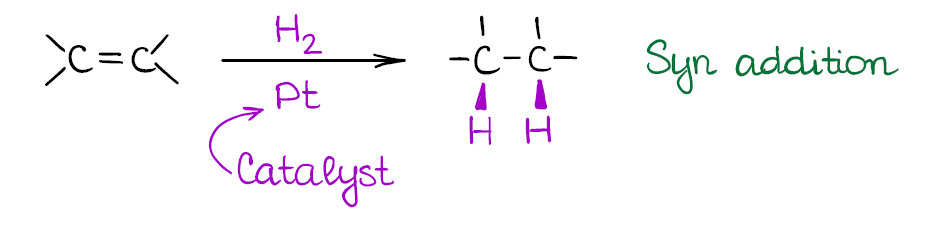
This is a type of a reduction reaction where you use hydrogen gas at a reasonably high pressure to get rid of the double bond. This reaction of alkenes happens on the surface of a metal catalyst. The typical catalysts for the alkene hydrogenation are the platinum (Pt), palladium (Pd), and nickel (Ni). Sometimes you’ll see the Pd/C instead of just the pure metal. This is, essentially, the same thing. Palladium gets electroplated on the carbon surface (a few atomic layers) making it a cheap alternative to the very expensive platinum group metal.
While catalytic hydrogenation of alkenes is a powerful reaction, it is a bit too powerful. This reaction will reduce all double or triple bonds to single bonds in your molecule! So, you should be very careful when using this method in your synthesis. There’s also no way to do a 1 equivalent of hydrogen, since you’re going to be using hydrogen gas at a high pressure (usually, at least several atmospheres).
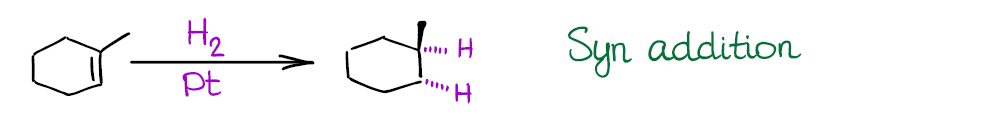
Also, the reaction is a syn addition that adds both hydrogens to the same face of the molecule. This is an important consideration as it may influence your synthetic decisions.
Halogenation of Alkenes
In this reaction you’re going to be adding two halogen atoms to your double bond:
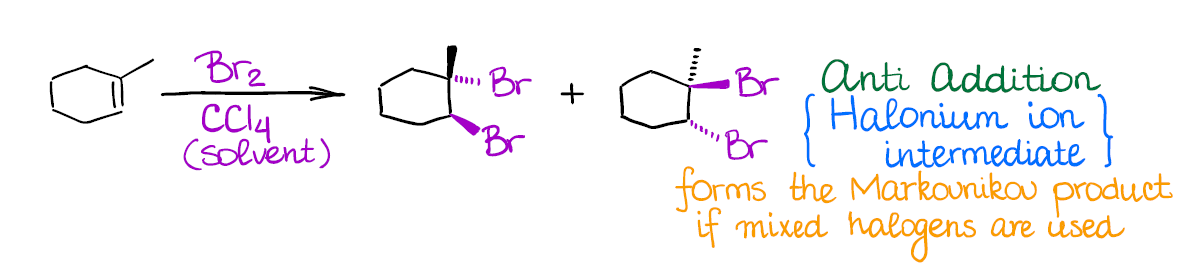
This reaction has a few important features:
- Halogenation of alkenes is an example of an anti-addition (stereospecific). This means that both halogen atoms will be adding to the carbons of the double bond in a trans fashion. Generally, you’re not going to have much stereoselectivity in this reaction, you’ll form a 50/50 mixture of two enantiomers.
- The intermediate in this reaction is a 3-membered ring that we call a “halonium ion.” Since there’s no carbocation in this reaction, you’re not going to see any carbocationic rearrangements. The halonium ion intermediate is also what makes this reaction a strict anti-addition.
The only tricky point in this reaction might be the use of mixed halogens. For instance, I-Cl, in which case you’ll form a Markovnikov product. Also, generally only chlorine and bromine are used in this reaction. Among all reactions of alkenes, this one is, probably, one of the most “iconic” and recognizable reaction.
Oxyhalogenation of Alkenes
This one is quite similar to halogenation in terms of the mechanism–you’re also going to be forming the halonium ion intermediate.
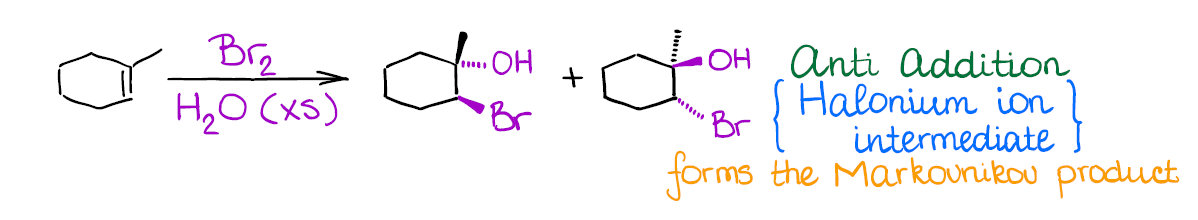
Interestingly, in this reaction you have a competition of two nucleophiles: bromide anion (that you form after the initial attack on the alkenes) and water. Since negatively charged bromide is a much better nucleophile than water, you need to play the statistics game here and use a large excess of water. This way, the chances of bromide acting as a nucleophile are statistically small. Thus, water is going to be the attacking species opening the bromonium ion.
An important thing to remember about this reaction is regioselectivity. You’re always going to form the Markovnikov product as the major product. Many reactions of alkenes have a certain stereochemistry and regiochemistry associated with them. Markovnikov regioselectivity means that the electrophile (the halogen here) going to end up on the less substituted carbon of the double bond, while the hydroxyl (-OH) will be on the most substituted carbon.
Alkoxyhalogenation of Alkenes
If you use a large excess of an alcohol instead of water in a reaction of an alkene with a halogen, you’re going to end up with an ether product instead of a halohydrin like in the previous reaction.
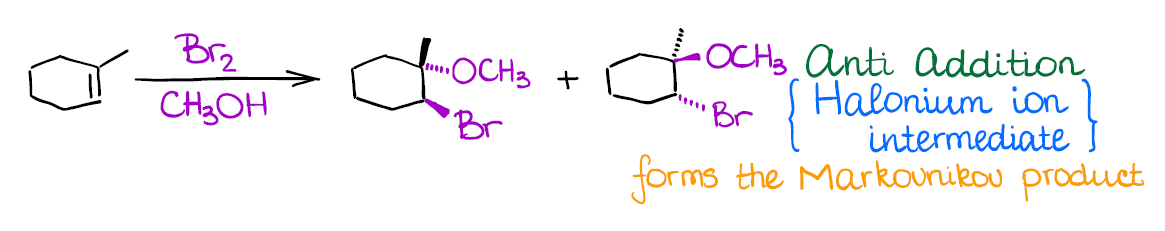
Mechanistically, the reaction is similar to the previous two examples. Like the other two, it also gives anti-addition stereospecificity, and gives you a Markovnikov product. A really cool thing about this reaction is that it allows you to form 5- or 6-membered cyclic ethers when the -OH is a part of the original alkene:
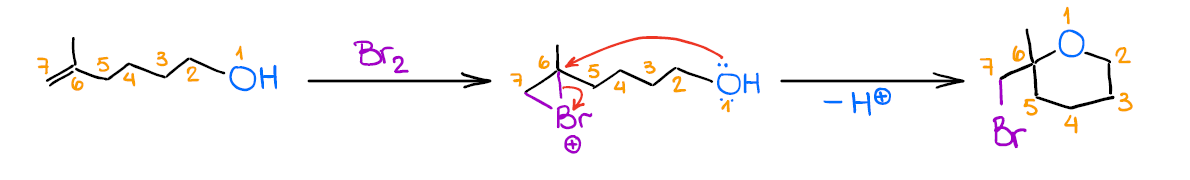
While this is not the most commonplace use for this reaction, it’s a cool trick to know for those extra points questions you may have on your test.
Hydrohalogenation af Alkenes
This is one of the most typical (and maybe even the first you’re going to learn) among the reactions of alkenes. Hydrohalogenation is regioselective and gives the Markovnikov product.
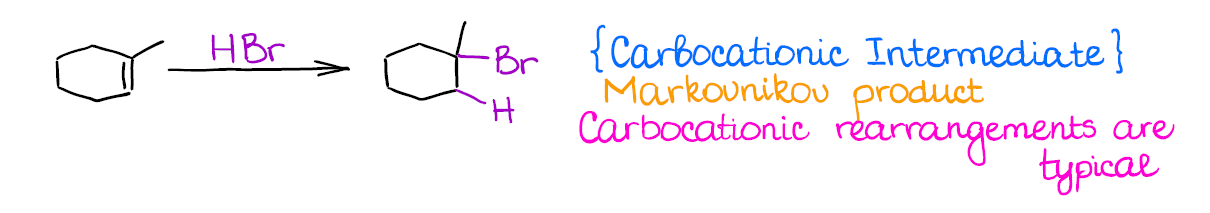
The most important thing about this reaction is that you’re forming a carbocation as an intermediate. And where you have carbocations, you have potential troubles! Carbocations will scramble the stereochemistry of any atom the “touch” in the mechanism. On top of that, they have a nasty tendency to rearrange, so your final product might be something very different from what you might’ve originally expected.
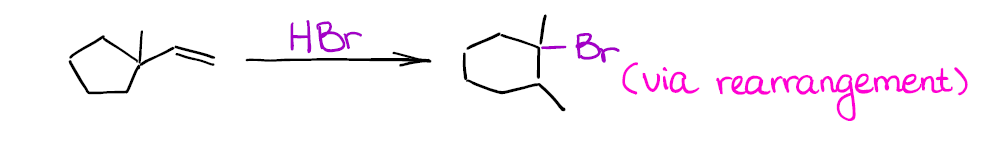
Can you draw the mechanism for this reaction to explain the structure of the final product? Hint: you’re going to have a ring expansion right after you form your initial secondary carbocation.
Catalytic Hydration of Alkenes
In this reaction you end up adding water to your alkene. Since water is not nearly acidic enough to protonate the double bond of an alkene by itself, you’ll need a strong acid as a catalyst.

You would typically see something like sulfuric acid (H2SO4) as a catalyst in this reaction. Make sure to never use HCl or HBr as a catalyst as those will just do the hydrohalogenation reaction!!!
Since this reaction also forms a carbocation as an intermediate, beware of possible rearrangements:
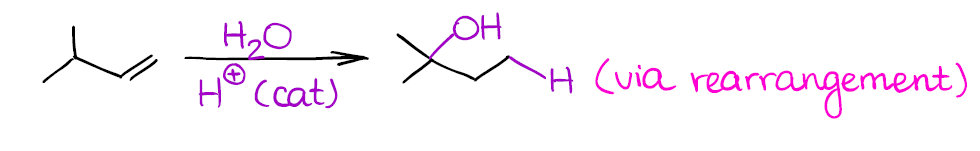
Remember, that if a carbocation can rearrange to give you a more stable carbocation, it will! So always check your reactions for any possible shifts or you might miss some points on the test.
Catalytic Addition of Alcohols to Alkenes
This reaction is very similar to the catalytic hydration.
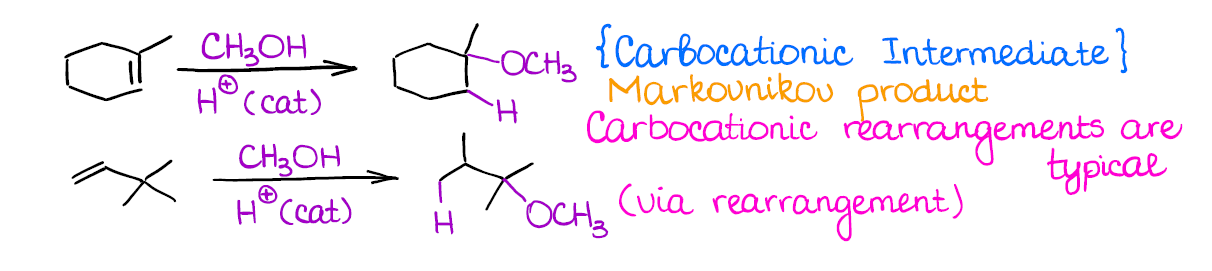
Just like in the case of the catalytic hydration, you’re going to form the most stable carbocation (Markovnikov rule), you’ll lose any stereochemistry, and you may have a rearrangement.
Oxymercuration-Demercureation (Reduction) of Alkenes
This is very useful reaction although you get to work with very poisonous mercury compounds. It gives the same product (alcohol) as the hydration of alkenes.
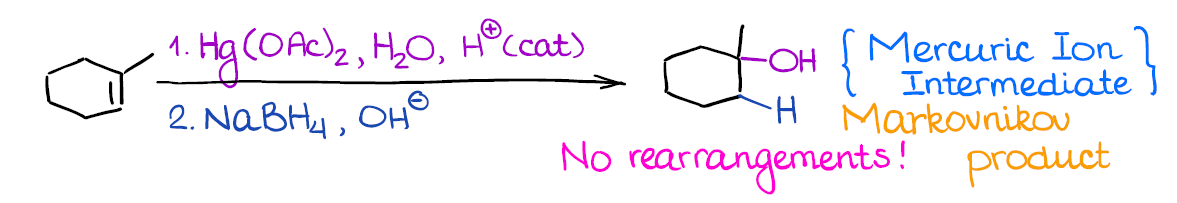
What’s really cool about this reaction, however, is that there’s no carbocation in this reaction! And no carbocation means no rearrangements! This is very useful when you’re dealing with sensitive or very strained compounds that have a high chance of rearranging.
Remember, this is a 2-step process, so it’s important to indicate that you have step 1 and step 2 next to your reagents above the arrow. Your instructor will understand what you mean if you don’t indicate 1. and 2., but some are picky about those small things.
Alkoxymercuration-Demercuration (Reduction) of Alkenes
This reaction is very similar to the regular oxymercuration. The only difference is that you’re using an alcohol instead of water as a nucleophile here.
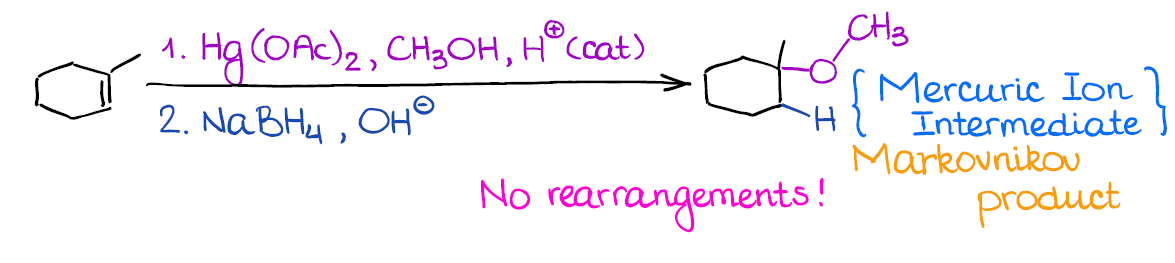
The huge benefit of this reaction, like with the oxymercuration, is the absence of the carbocationic intermediate, which means no rearrangements. This way, you can form a fancy ether without any fear of changing the carbon skeleton or ending up with your alkoxy group in a wrong place. You can even make cyclic ethers when you have -OH and C=C in the same molecule. Just remember, that this reaction can only reliably make 5- or 6-membered rings.
Hydroboration-Oxidation of Alkenes
This reaction is a reliable way of introducing the hydroxyl (-OH) group into your molecule in an anti-Markovnikov fashion.
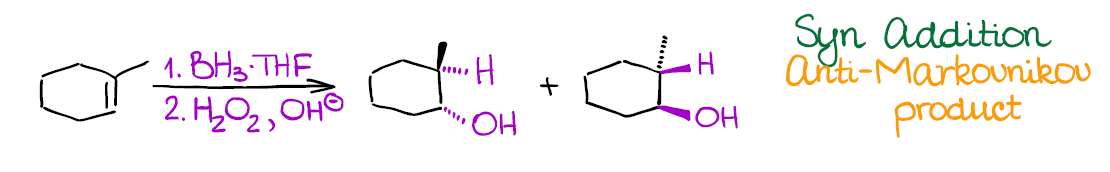
Hydroboration-oxidation has a couple of very important features:
- As I’ve mentioned above, it adds the -OH in the anti-Markovnikov way. Reaction also shows no carbocations in the mechanism, so you don’t need to worry about any rearrangements.
- This reaction is an example of a syn addition. This means that both -H and -OH will end up on the same face of the molecule cis to each other. This is a potentially useful (or problematic) for synthetic purposes and you can bet your instructor will test that!
Like the previous examples, this is a 2-step process. So, make sure to indicate it properly when writing it out!
Epoxidation of Alkenes
Epoxides are kind of a big deal in organic synthesis. So, it’s a good idea to know how to make them. One of the most common ways of making epoxides is the direct epoxidation of alkenes with peroxy acids. The nature of the peroxy acid is majorly irrelevant though. One of the most common peroxy acids used in this reaction is mCPBA (meta-chloroperbenzoic acid).
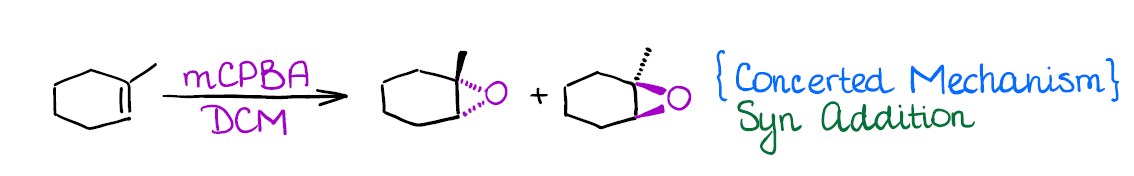
Epoxides have a very rich chemistry and you’ll cover those later in the course.
Ozonolysis of Alkenes
Ozonolysis is a very popular reaction on the tests. In the 15 years of me tutoring organic chemistry I have never seen an exam without one! What does ozonolysis do? It, in a nutshell, cuts through your double bond. This is why sometimes you can hear that this reaction is called the “cleavage” of a double bond.
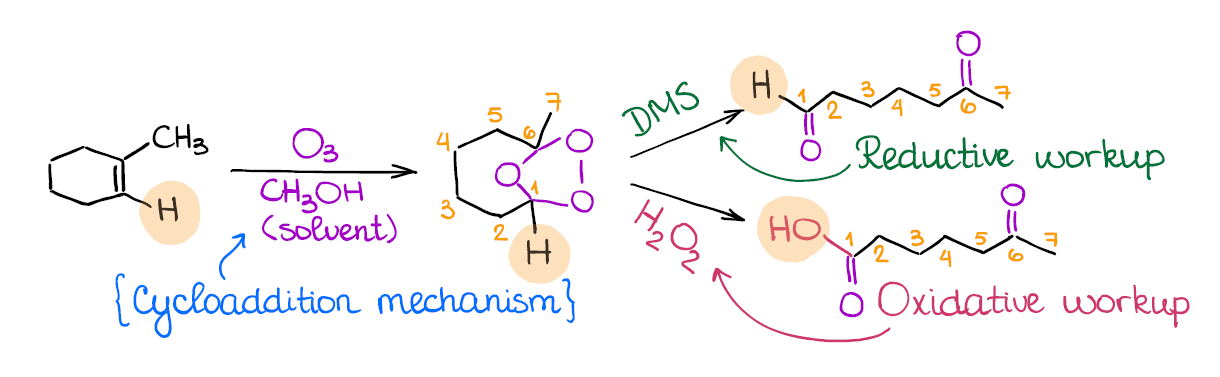
The important thing to remember about the ozonolysis is that it’s not one, but two different reactions. We have a reductive ozonolysis, and we have oxidative ozonolysis. While occasionally it won’t matter, most of the time these two will give different products. Reductive ozonolysis gives aldehydes and ketones, while the oxidative ozonolysis yields ketones and carboxylic acids instead of aldehydes.
How do you know which of the two you’re dealing with? The secret is in the second step. The reductive ozonolysis uses DMS (dimethyl sulfide) or Zn/HCl, while the oxidative ozonolysis will have the aqueous hydrogen peroxide (H2O2) in the second step.
Many instructors love using ozonolysis as a part of a complex puzzle-like questions where you need to identify a compound based on the ozonolysis fragments! So this reaction is definitely a must-know for anyone who wants to get an A!
Dihydroxylation of Alkenes
In this reaction you will be adding two hydroxyls (-OH) groups to your double bond at the same time. There are a couple of ways how dihydroxylation is done: one with osmium oxide (OsO4), and the other one with potassium permanganate (KMnO4).
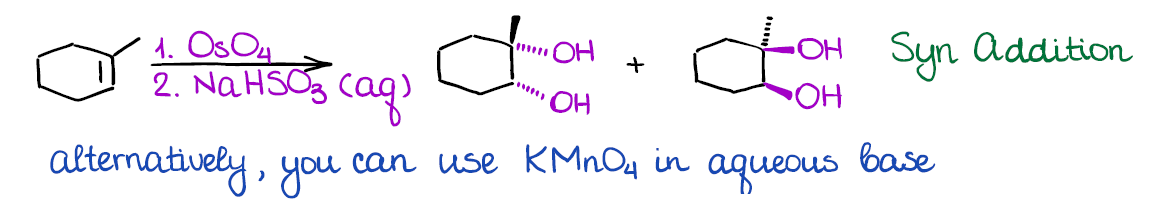
The advantage of the osmium oxide is that this reaction is extremely selective and gives good yields. The problem with osmium oxide is that it’s very toxic and may cause instant blindness upon exposure. So, it should be handled with extreme care and in small quantities only. There are many modifications to this reaction that only use osmium oxide in a catalytic quantity making it way safer.
The alternative method uses potassium permanganate. While that reagent is significantly safer to work with than osmium oxide, it is a very strong oxidizing agent. Thus, you may often over-oxidize you compound by further oxidizing the resulting alcohol. From the synthetic perspective, potassium permanganate is very rarely used in actual lab. However, many instructors like to show this reaction as it appears on MCAT and other standardized tests.
Cyclopropanation of Alkenes
There are two common methods that you may see in your course. The first one is called the Simmons-Smith reaction.
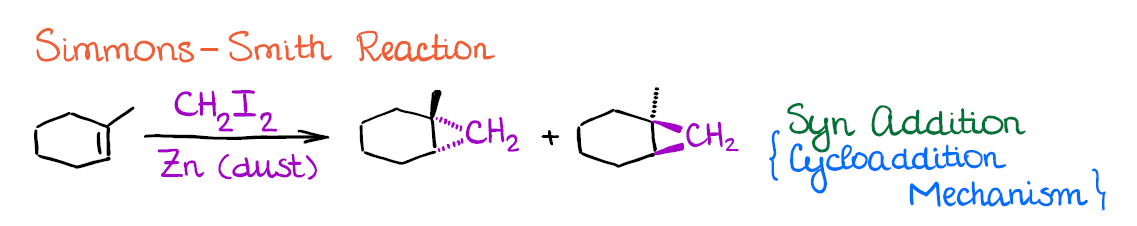
Simmons-Smith is a neat reaction that can easily convert any C=C bond into a cyclopropane. While it has a very limited synthetic application, this is still a popular reaction to cover in the alkenes unit, so it’s a good one to know.
The other cyclopropanation method uses dichloromethane that is formed in situ through the reaction of chloroform and a strong base like tert-butoxide.
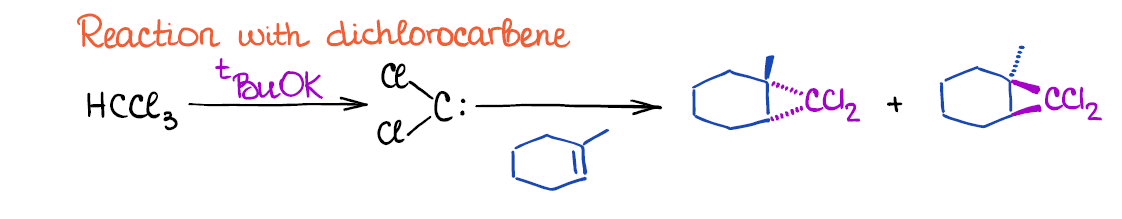
Dichlorocarbene is a very reactive 6-electron carbon species so it has to be generated in the same reaction mixture where the double bond is already present. You cannot isolate it and keep it for later use.
Radical Hydrohalogenation of Alkenes
This is a very different reaction from anything you’ve done before as it has a radical mechanism. It’s also another method how you can introduce a functional group in an anti-Markovnikov fashion.
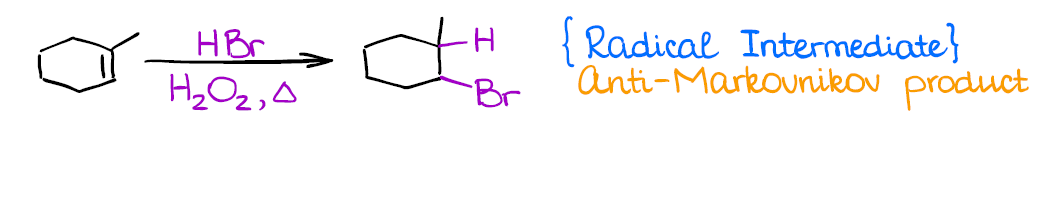
Similar to carbocations, you’re going to lose stereochemistry and make all possible stereoisomers. Radicals, unlike carbocations, don’t tend to rearrange.
So, that was just a brief overview of the reactions of alkenes. I’ve got a detailed set of notes on reaction of alkenes describing all these reactions, going over each mechanism and all important aspects of each reaction.