Allylic and Benzylic Reactivity
In this tutorial, I want to talk about allylic and benzylic reactivity—and why compounds with these molecular moieties hold a special place in organic chemistry.
What is so special about the benzylic and allylic positions?
Probably the most important thing to keep in mind when thinking about allylic and benzylic positions is resonance stabilization. If we look at the allylic carbocation, we’ll see resonance stabilization involving two roughly equally important resonance contributors. This gives us an overall hybrid with a partial positive charge distributed across two positions on the molecule.

When it comes to the benzylic position, resonance becomes significantly more extensive. We’ll see a larger number of resonance structures, which ultimately means that benzylic positions—whether we’re talking about carbocations, carbanions, or radicals—are much more stable than their allylic counterparts.
Another important thing to keep in mind about the benzylic carbocation is that the resonance contributor with the intact aromatic ring is always the major one. In contrast, in allylic systems, all resonance contributors usually have roughly equal importance.
Now, we’re going to be dealing with a lot of resonance throughout this tutorial. However, I’m not going to be drawing out every single resonance structure. If you need a refresher on how resonance works, I have a tutorial on that—check that one out first. For the rest of this tutorial, I’ll assume you’re comfortable with resonance and can draw the contributors when needed.
Substitution in Allylic Halides
So, let’s look at the chemistry of allylic positions, starting with SN1 reactions. Here’s a fairly typical SN1 reaction, where I have a tertiary alkyl halide reacting with water, ultimately giving an alcohol as the final product. We know that in unimolecular reactions like SN1, we form a carbocation intermediate—so the stability of that carbocation is extremely important.

Now, let’s look at a very similar reaction—but this time involving a tertiary allylic alkyl halide. Here’s where things get interesting: the carbocation is stabilized by resonance. Because of that, we’re going to end up with two different products. One of them is written out here—it looks very similar to the product from the previous reaction. The other one comes from the alternative resonance contributor, giving us this second product. These two products are constitutional isomers—also called structural isomers—of each other.
So, whenever you’re dealing with SN1 reactions involving allylic positions, you have to be very careful, because resonance can lead to multiple products. Another thing to note here: due to resonance stabilization of the carbocation intermediate, the reaction proceeds significantly faster. In fact, SN1 reactions involving allylic carbocations can be over 100 times faster than comparable reactions with non-resonance-stabilized carbocations.
Substitution in Benzylic Position
Now, when it comes to reactions involving the benzylic position, we see a similar pattern in terms of resonance stabilization. However, due to the much greater stabilization of the benzylic carbocation, the reactivity is even more pronounced. These benzylic intermediates are incredibly reactive—and can react hundreds of times faster than non-stabilized carbocations.

But before we move on to the next reaction, I want to point out that since we’re dealing with resonance here, the substituents on the aromatic ring matter a lot! For instance, let’s look at this first reaction where I have a benzylic halide, and the aromatic ring has a methoxy group attached to it. Now, let’s compare this reaction to a similar-looking molecule—but this time the aromatic ring has a nitro group instead.

If we compare the reaction rates of these two cases, the first one proceeds incredibly fast, while the second one is dramatically slower. The reason is: in the first case, the methoxy group is an electron-donating group. It helps stabilize the carbocation intermediate. In the second case, we have a nitro group, which is an electron-withdrawing group—and that destabilizes the positive charge on the carbocation, making its formation less favorable. So, whenever you’re dealing with the benzylic position and you have a carbocation, carbanion, or radical at that site, always double-check what kind of groups are attached to the aromatic ring. That way, you can recognize the electronic effects those groups exert—and whether they’ll stabilize or destabilize the intermediate and the reaction overall.
Elimination Reactions in Allylic and Benzylic Halides
All right, moving on to E1 reactions—here’s a fairly typical example where I have an allylic alcohol reacting with sulfuric acid at high temperature. Reactions like this tend to give conjugated systems as the final product, due to the fact that conjugated systems are well stabilized by delocalization. But the issue with these types of molecules is that they tend to undergo easy polymerization under acidic conditions. That means E1 reactions involving allylic and benzylic compounds are generally a bad idea and don’t give good yields—so we won’t spend more time on them.

However, when it comes to E2 reactions, things become a bit more interesting. Let’s look at a typical example you might see on an exam: we have a molecule with a primary halide, and an aromatic ring sitting on the second carbon. If we analyze this reaction using a basic predictive model, here’s what we see: sodium ethoxide acts as a base and a nucleophile. Our leaving group is on a primary carbon. So normally, we’d expect this reaction to proceed via the SN2 mechanism.
Experimental data shows that about 95% of the product comes from an E2 reaction—not SN2. So, why might that be the case? One reason is that benzylic protons are significantly more acidic than typical alkyl protons. Here, we’re looking at a pKa of around 45, compared to ~55–60 for standard alkyl protons. The real explanation lies in the molecular orbitals.
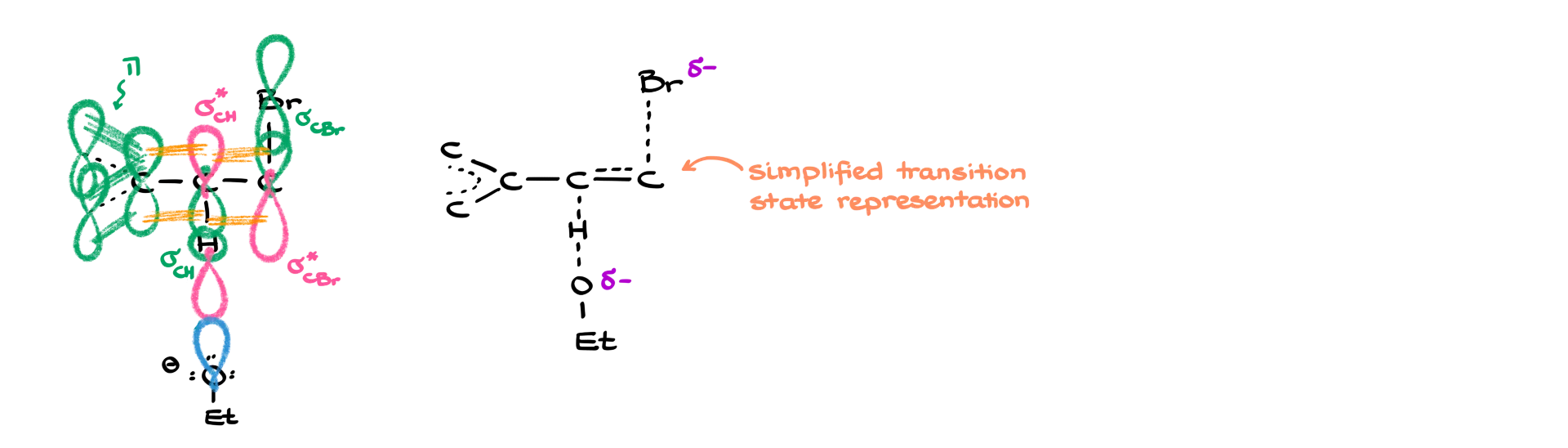
Here’s a molecular orbital representation of the transition state for this reaction. The important thing to recognize is that the π orbitals of the aromatic ring—or an adjacent double bond—can stabilize the transition state. This happens through orbital overlap with the σ* (sigma antibonding) orbital of the C–H bond that’s breaking. That stabilizing interaction lowers the energy of the transition state. As a result, the E2 elimination pathway becomes significantly more favorable and proceeds faster. And finally, another argument in favor of elimination here is that it gives a conjugated product—which is thermodynamically more stable.
SN2 Reactions in Allylic and Benzylic Positions

Now, moving on to SN2 reactions—things get pretty interesting here as well. Let’s look at three comparable reactions, starting with Reaction A, where I have a n-propyl bromide reacting with a thiolate ion, giving the corresponding sulfide. Next, Reaction B involves an allyl bromide undergoing the same reaction. And finally, in Reaction C, we have benzyl bromide doing the same thing.
If we compare the reaction rates and take Reaction A as our baseline, we’ll see that Reaction B proceeds about 70 to 100 times faster. And Reaction C—with the benzylic position—is over 100,000 times faster than Reaction A. But since SN2 reactions don’t involve intermediates, we know this must be about the transition state.
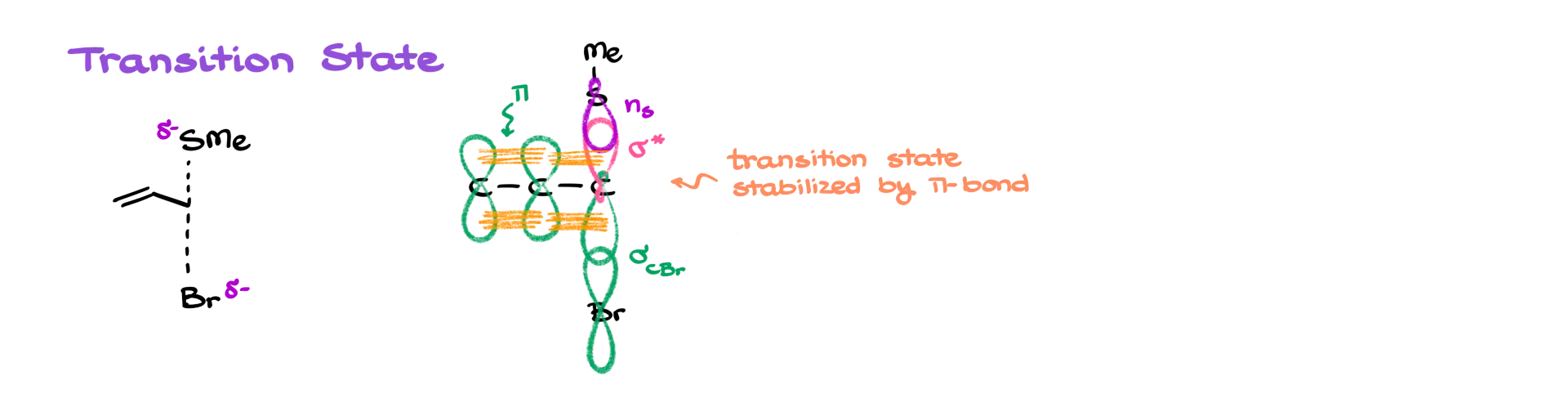
Just like we saw earlier, the adjacent π system—whether it’s a double bond or an aromatic ring—can stabilize the transition state. That stabilization happens through orbital overlap, which lowers the energy of the transition state. And when the transition state is lower in energy, the reaction becomes much more favorable—and much faster.
Allylic and Benzylic Carbanions
Next, let’s talk about allylic and benzylic carbanions. In the case of benzylic anions, electron-withdrawing groups on the aromatic ring help stabilize the negative charge. For allylic anions, resonance stabilization is usually more limited. Also, we typically don’t see free carbanions just sitting on carbon atoms—they’re not stable on their own. That’s why allylic and benzylic anions usually show up as part of organometallic compounds, like Grignard reagents or organolithiums. And that’s where things get interesting.

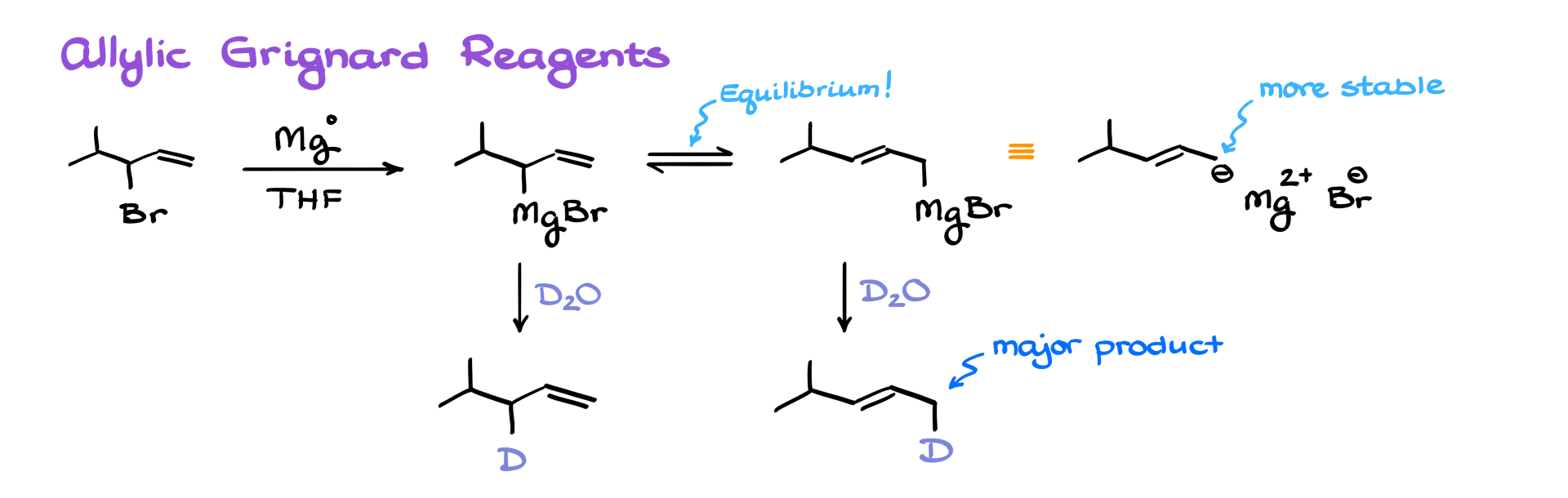
Let’s look at an allylic Grignard reagent. Say I start with this allylic bromide. I react it with magnesium in the presence of THF or anhydrous ether—standard conditions for forming a Grignard reagent. As a result, I’ll get the corresponding allylic Grignard reagent. But here’s something interesting about these compounds. Due to resonance stabilization of the negative charge, the allylic Grignard reagent exists in a fast equilibrium. In this equilibrium, the magnesium ion shifts between carbons 1 and 3 of the allylic system. And importantly, the form where the negative charge is on the less substituted carbon is generally more stable. So, if we react this organometallic compound with something like D₂O—or even just regular water—to quench it, we’ll end up with two different products. The major product will be the one that comes from the less substituted carbanion contributor.
Benzylic and Allylic Radicals
Now, moving on—I want to talk about benzylic radicals.
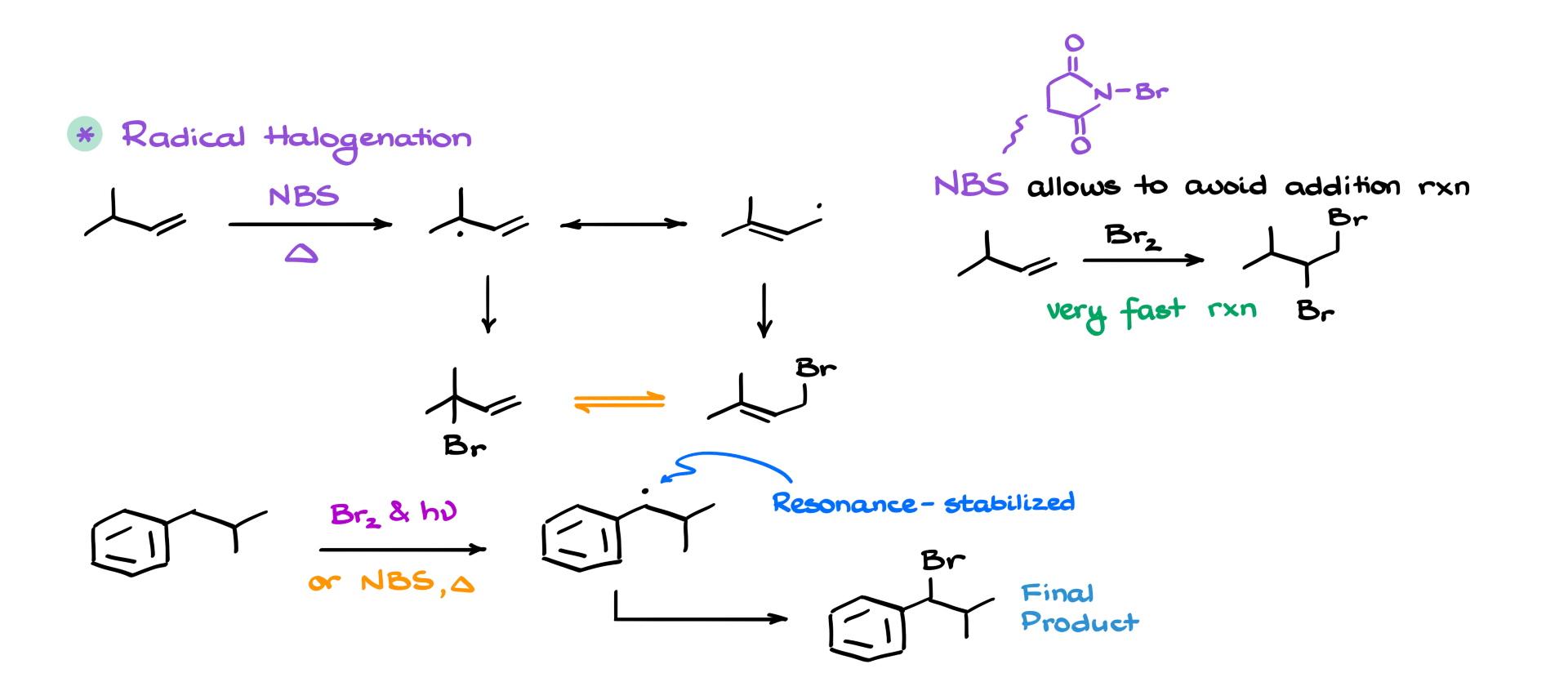
One of the important reactions involving allylic and benzylic systems is radical halogenation. For instance, let’s say we have this allylic system here, and we’re reacting it with NBS—that’s N-bromosuccinimide. As a result, we’ll form the corresponding allylic radical, which has the following resonance contributor. Because this radical has two different resonance forms, we’ll end up with two different products. And importantly, because the leaving groups can dissociate easily, these two molecules are also in equilibrium with each other.
I’m not going to get into the full mechanism here, but one key point: NBS helps avoid direct addition of bromine to the double bond—so it keeps the reaction selective. Now, when it comes to radical halogenation of the benzylic position, things are even easier. You can use bromine, NBS, or pretty much any radical halogenation method you like. Just like in the allylic case, you’ll form a resonance-stabilized radical at the benzylic position, which gives you the final halogenated product. The great thing about benzylic radical reactions is that they’re typically clean. The aromatic ring stabilizes the intermediate, and it stays intact throughout the reaction. So you don’t get a bunch of side products muddying up your mixture.
Oxidation of Allylic and Benzylic Positions
We also have a unique reaction for allylic and benzylic alcohols: selective oxidation using manganese dioxide.

Mechanistically, I’ll abbreviate the R group as “allylic or benzylic substituent” since the reaction works the same way for both. In the first step, we bring in manganese dioxide, and the alcohol oxygen attacks the manganese center. After a couple of proton transfers, we get this intermediate. From there, we have a hydrogen abstraction step, forming a biradical species. That leads to our final product: the corresponding aldehyde. The awesome part is that this reaction is selective—it only oxidizes allylic and benzylic alcohols, and it leaves all other alcohols in your molecule untouched.

And finally, I can’t wrap up this tutorial without talking about benzylic oxidation. If we have an aromatic compound with a benzylic position that has at least one hydrogen, we can oxidize it using potassium permanganate or potassium dichromate in sulfuric acid. That gives us the corresponding carboxylic acid. And the cool part? It doesn’t matter what else is on that benzylic carbon—everything gets chopped off, and you always end up with the benzoic acid derivative.
So let’s look at a couple of examples. In the first molecule, I’ve got an aromatic ring connected to another ring—but that second ring doesn’t matter. What matters is that we have a hydrogen on the benzylic carbon. That allows us to oxidize it and chop off the rest of the side chain, giving us the corresponding dicarboxylic acid—in this case, phthalic acid. In the next example, I’ve got two groups on the aromatic ring. Only one of them has a benzylic hydrogen—so only that group will undergo oxidation. The tert-butyl group on the bottom has no benzylic hydrogen, so it stays untouched. The final product in this case is a mono-carboxylic acid.
These types of questions are common on exams—but the great part is, you don’t need to know the full mechanism.