Dess-Martin Periodinane and Dess-Martin Oxidation
When it comes to the oxidation of primary alcohols to aldehydes, there are three classic methods that we normally talk about. These are oxidation with PCC or PDC, the Swern oxidation, and of course the Dess–Martin oxidation, which is the youngest of the three and probably one of the best.
For the most part, these three methods are interchangeable. However, each one has its own benefits and drawbacks. In this tutorial we are going to talk about Dess–Martin periodinane, how we make Dess–Martin periodinane, and the reactions that this compound can undergo, mainly focusing on oxidation chemistry.
Synthesis of the Dess-Martin Periodinane
To make Dess–Martin periodinane, we start with 2-iodobenzoic acid, like the compound shown on the screen. This is then oxidized with a stronger oxidizing agent, things like bromate, oxone, or other reagents capable of taking iodine all the way to the +5 oxidation state.
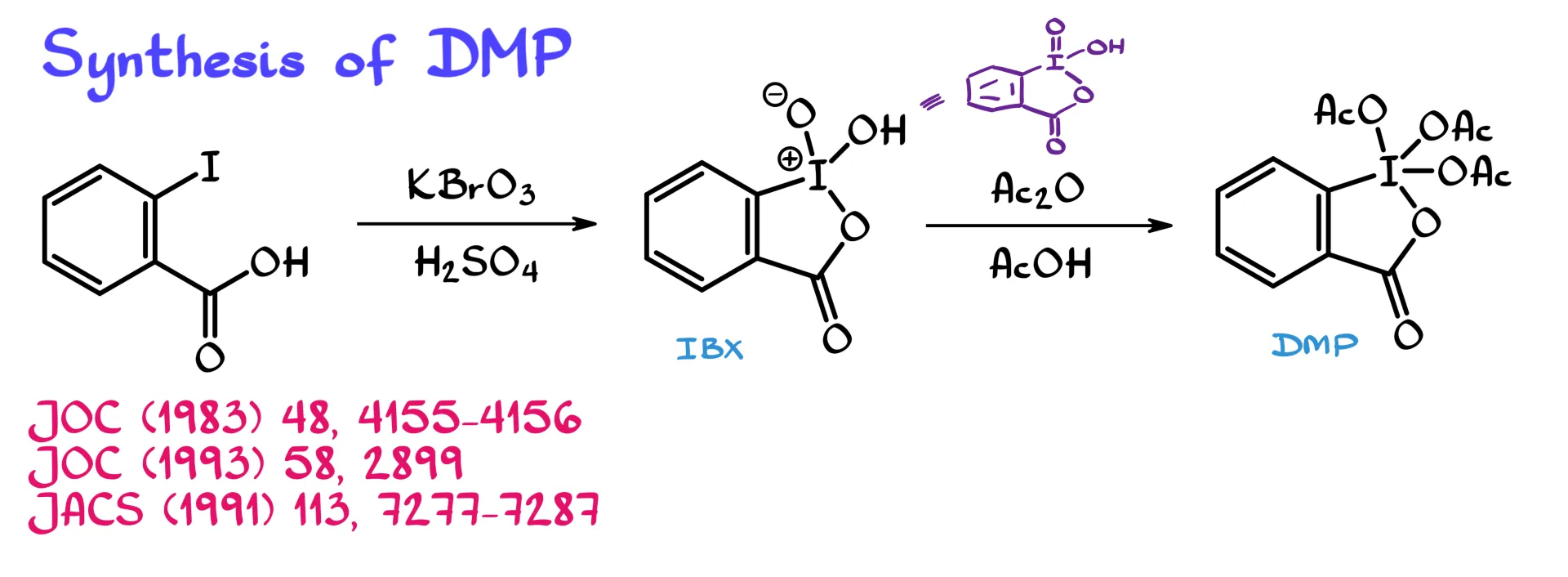
What I have drawn here as the intermediate is iodoxybenzoic acid, often abbreviated as IBX in the literature. This compound has been known for well over 100 years, but it has a lot of problems when it comes to its chemistry. One of the biggest problems is that it is really difficult to dissolve in almost anything. And if we cannot dissolve the compound, we cannot really do much chemistry with it.
The way I drew this compound shows the zwitterionic form, with a negative charge on oxygen and a positive charge on iodine. That is one of the two common ways you will see this reagent written. You could also see IBX drawn in the alternative neutral form, and you could even think of those as resonance contributors if you like.
For the purposes of this tutorial, whenever I come across a compound like this, I will simply call it iodoxybenzoic acid. IBX is not actually Dess–Martin periodinane, so to convert it into the final periodinane we need to acetylate it. That gives us DMP, Dess–Martin periodinane.
Oxidation of Alcohols with DMP
For this tutorial, as I mentioned, we are going to focus on the oxidation ability of DMP and its ability to convert alcohols into the corresponding aldehydes or ketones.
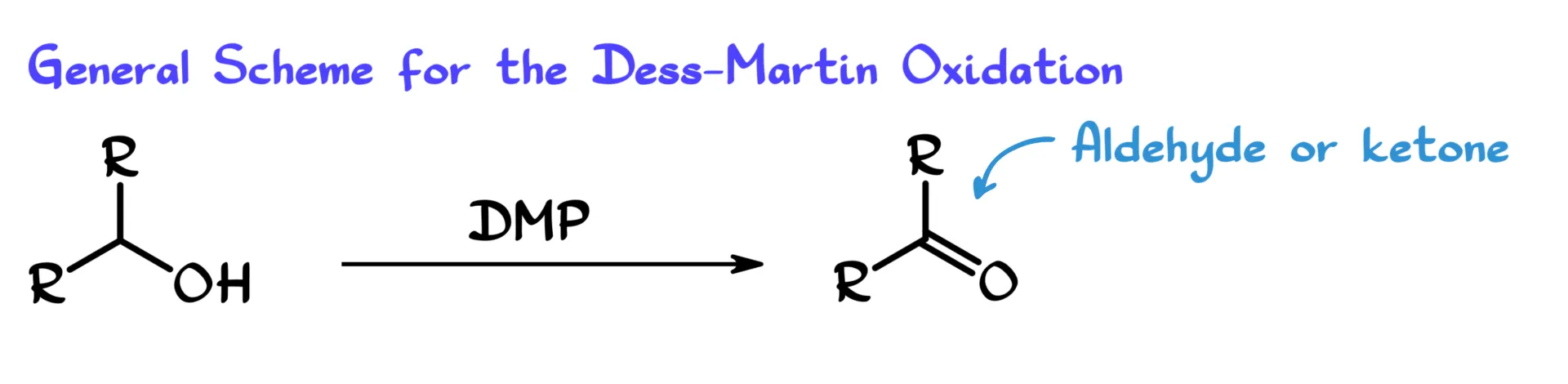
If we are working with a primary alcohol, so one of those R groups is just hydrogen, we are going to end up with an aldehyde as the final product. Of course, if we have a secondary alcohol like the one in this generic scheme, we are going to end up with the corresponding ketone. That part is a little less interesting because no matter how you oxidize a secondary alcohol, you are going to get a ketone anyway, so that is kind of the boring part.
Why Use DMP
Before we go into the oxidation mechanism itself, I want to spend just a few moments discussing why we might want to use DMP instead of Swern oxidation or PCC.

First, the reaction is typically quite fast. We are usually talking about 30 minutes to 2 hours, while reactions with something like PCC can easily take 12 hours or even more.
The reaction also does not usually require a large excess of oxidizing agent, unlike PCC, where that excess can be quite large.
And finally, unlike Swern or especially PCC, this reaction gives a very clean workup and typically proceeds under fairly neutral conditions. So if you have a sensitive compound, it is more likely to survive the process.
If you have ever done a PCC oxidation in the lab, you know that the reaction gives dark, messy mixtures and is a nightmare to work up. Not to mention, you then have to figure out what to do with the rather toxic chromium used in the reaction. And when it comes to Swern oxidation, let’s just say that reaction is stinky, and that is a compliment.
Mechanism of the Dess-Martin Oxidation
In the very first step, we take our primary alcohol. For simplicity I am using 1-butanol here, and we treat it with our DMP reagent.
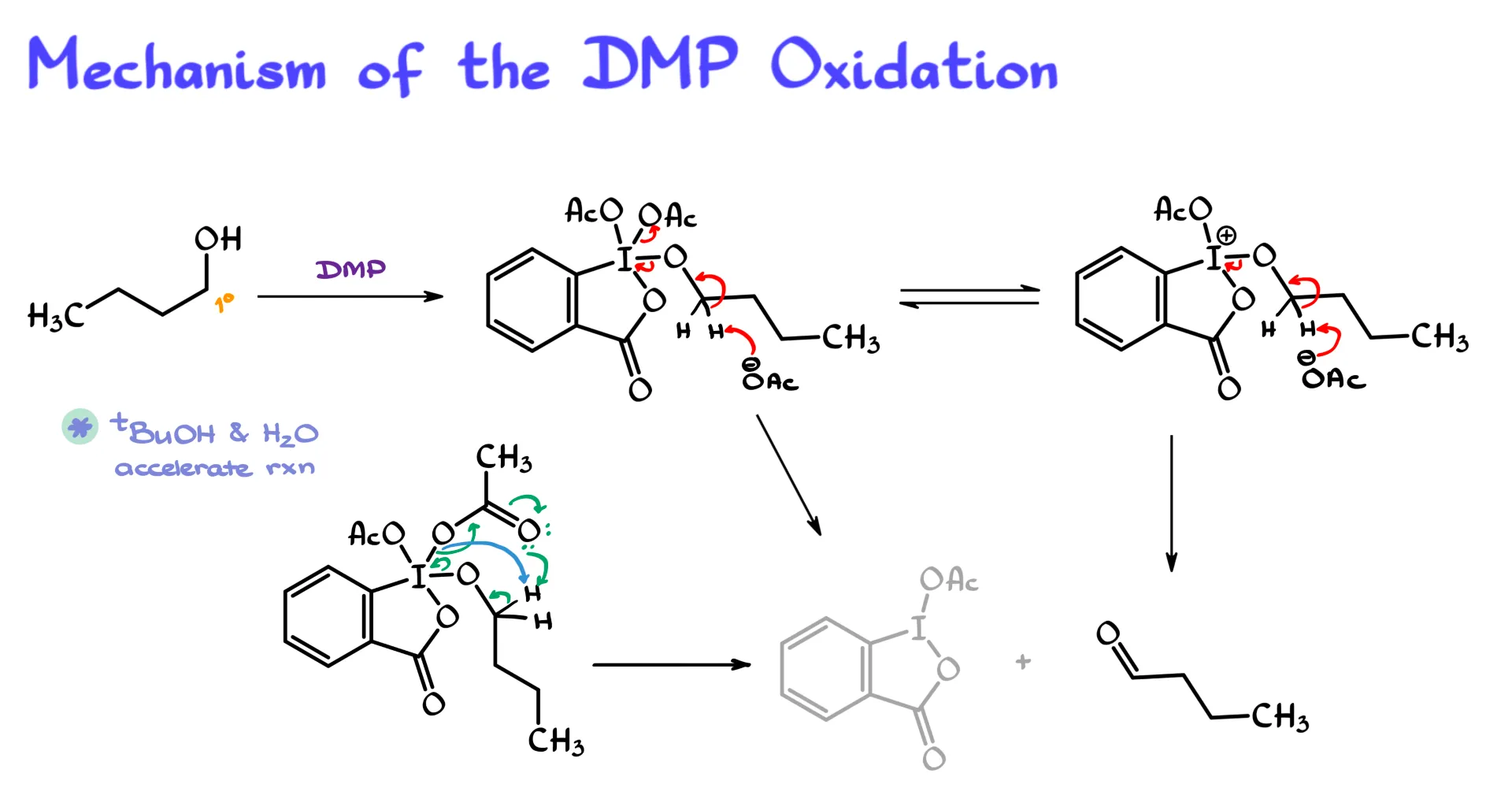
Since the acetate groups on iodine can more or less freely dissociate, we can replace one of them with our alkoxide. So now we have a structure where one acetate has been replaced by the oxygen of the alcohol, which is now connected to the rest of the butyl chain.
This intermediate exists in a rapid equilibrium where one of the acetate groups can dissociate and float around in solution. From here, there are a couple of possible mechanisms. They are not all that different, they are actually quite similar, but they are still worth mentioning.
In the first version, an acetate ion, or essentially any suitable conjugate base present in solution, removes one of the α hydrogens next to the oxygen. The electrons go onto oxygen, then from oxygen onto iodine, and one of the acetate groups leaves. This step releases the aldehyde as the final organic product and gives an iodine-containing byproduct in which iodine is now in the +3 oxidation state.
The other version starts from the molecule on the right, where one acetate has already dissociated. The same basic thing happens. Acetate comes in, removes the proton, the electrons move toward iodine, and in this case there is simply one fewer group to expel because the acetate had already dissociated beforehand.
There is also evidence that this reaction can happen intramolecularly without a free acetate floating around. We still target the same α hydrogens, but the internal oxygen or acetate-like group can participate directly in removing the proton, followed by the same flow of electron density. In the end, we get the same products.
Another version that has also been reported in the literature involves a slightly different internal proton transfer, but again it leads to the same general outcome. Acetate leaves, the aldehyde is formed, and iodine is reduced to the +3 oxidation state.
The typical textbook mechanism is the first one I described, and that is probably the one you are most likely to use in class. But I have seen the second version presented as well. And as I mentioned, the literature also supports an intramolecular pathway. So which variation you use really depends on what your instructor wants.
Interestingly, methanol, t-butanol, or even water in some cases can significantly accelerate this reaction. The reason is that the alcohol can replace another acetate in the complex, increasing the concentration of acetate in solution and sometimes speeding up the reaction dramatically. That is actually pretty neat, because neither Swern oxidation nor PCC can tolerate aqueous conditions or other alcohols nearly as well.
This makes Dess–Martin periodinane a very powerful oxidation technique that works under many different conditions and tolerates a variety of functional groups. From a synthetic standpoint, it is extremely versatile.
On top of that, Dess–Martin periodinane can be stored on the shelf for quite a long time. Swern oxidation intermediates are not storable, and PCC itself is not particularly stable either because it is so hygroscopic. It pulls moisture out of the air and gradually ruins the reagent. So in most cases PCC, and definitely the Swern oxidation intermediates, have to be prepared right before the reaction, which makes things more complicated.
Notable Side-Reactions
There are a couple of notable side reactions that I want to mention, because you need to be aware of them. One is the reaction with vicinal diols. If we take a vicinal diol and react it with DMP, one significant intermediate can be a cyclic species where both alkoxy groups are bound to the iodine, giving this cyclic intermediate.
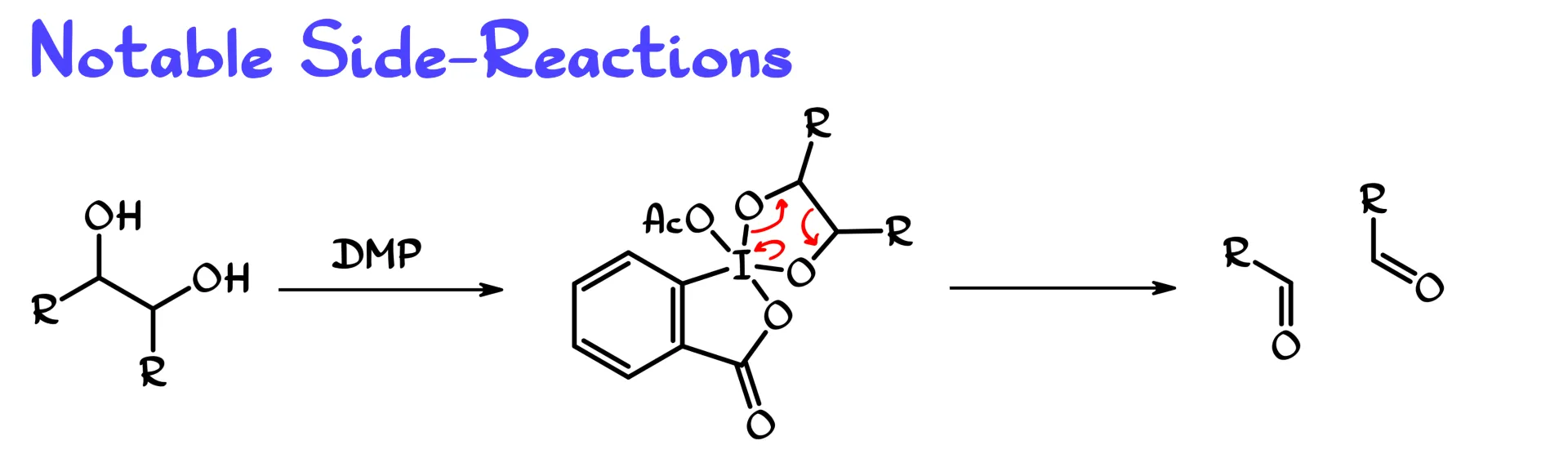
From this point, similar to other hypervalent iodine compounds, we can get an oxidative cleavage of the carbon–carbon single bond between the two oxygen-bearing carbons. That gives two carbonyl compounds and breaks the bond between them.
Depending on the nature of your vicinal diol, or pinacol if you want to think of it that way, this pathway can be either a minor inconvenience or the major reaction pathway. So if you are proposing a Dess–Martin oxidation of a vicinal diol, you definitely need to be aware of this complication because it could, in principle, completely ruin your molecule.
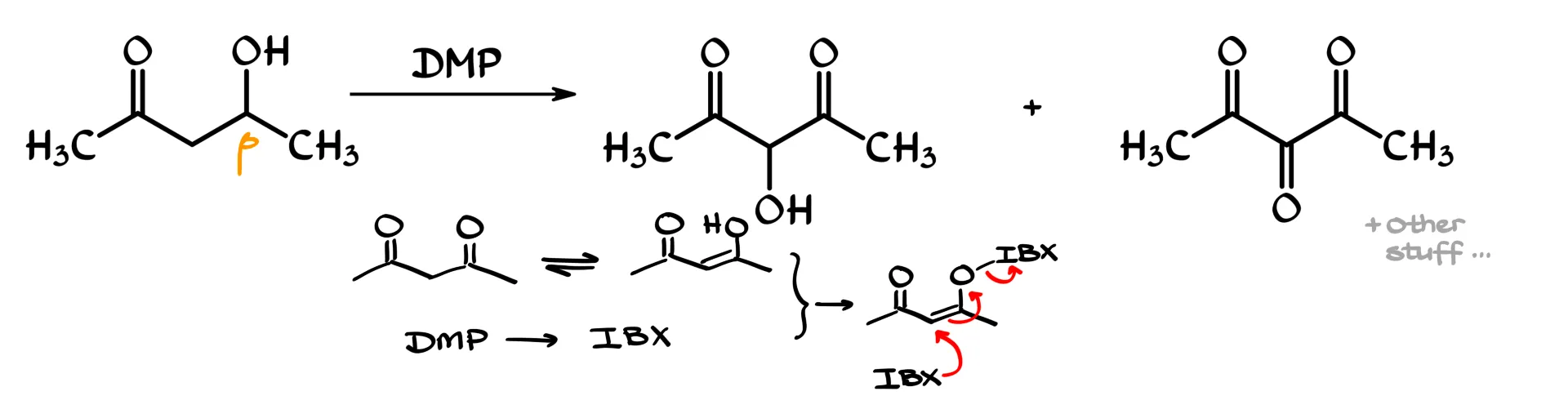
Another interesting side reaction shows up with β-hydroxy carbonyl compounds. In some cases, instead of simply getting the expected oxidation product where the OH group becomes a C=O bond, you can also see products where an OH or even another carbonyl appears at the α-position, right in the middle of the molecule.
This can be avoided by carefully controlling the reaction conditions. Under aqueous conditions, DMP can be hydrolyzed back to IBX, and those two species can interact to form a highly reactive intermediate that I will show very generically. This intermediate behaves like an activated enol, and it can react with another equivalent of IBX. In effect, you get oxidation while also introducing oxygen at the α-position. From there, depending on how the reaction proceeds, you can end up with either an OH group or even a carbonyl at that central carbon.
These side reactions do show up if the reaction is not done carefully, or if the conditions are poorly optimized. But despite that, DMP is still one of the best oxidation techniques available for primary alcohols, and for secondary alcohols as well.
So when you are putting together a multistep synthesis, I would strongly encourage you to use DMP rather than PCC or Swern oxidation whenever it makes sense.