Enols and Enolates
This section has everything you’ll need to know about the chemistry of enols and enolates. Here’s a brief intro to what enols and enolates are all about.
Acid-Promoted Enolization
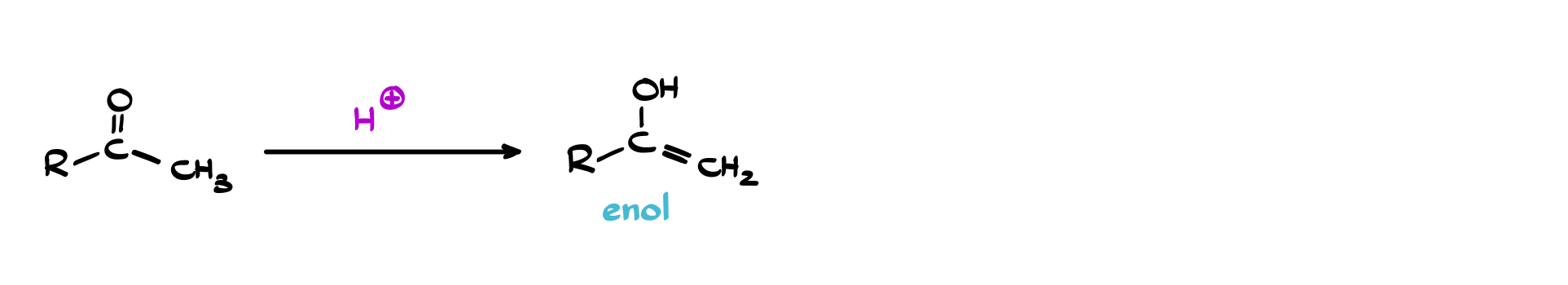
If you take a carbonyl compound and move one proton and one π bond around, you get what we call an enol: a molecule with a C=C double bond and an –OH group attached directly to it. This process is called keto–enol tautomerism, and while the keto form is usually more stable, the enol is far from irrelevant. It’s reactive in ways that the carbonyl itself is not, particularly because that C=C bond is electron-rich and ready to engage in electrophilic reactions.
Base-Promoted Enolization
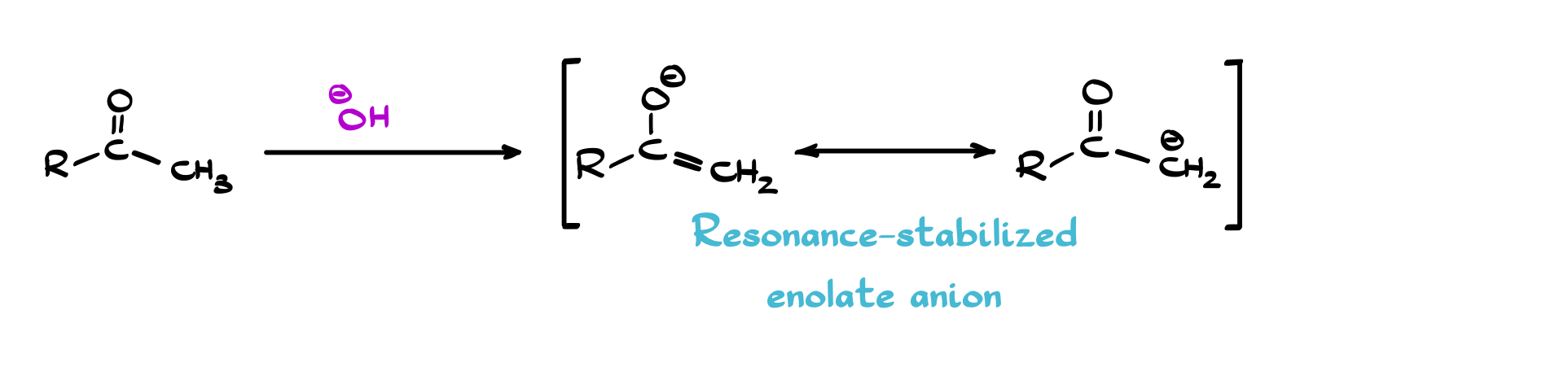
Now, if you take it one step further and deprotonate the α-carbon, you get an enolate—and this is where things really become useful. Enolates are resonance-stabilized anions, with electron density shared between carbon and oxygen. That dual personality is the key: sometimes they behave like a carbon nucleophile (forming C–C bonds), and sometimes like an oxygen nucleophile. Most of the carbon–carbon bond-forming reactions you learn in organic chemistry: aldol reactions, Claisen and Dieckmann condensations, alkylations are all built on this simple idea. If you can form an enolate, you can start building molecules in a controlled and predictable way.
Halogenation
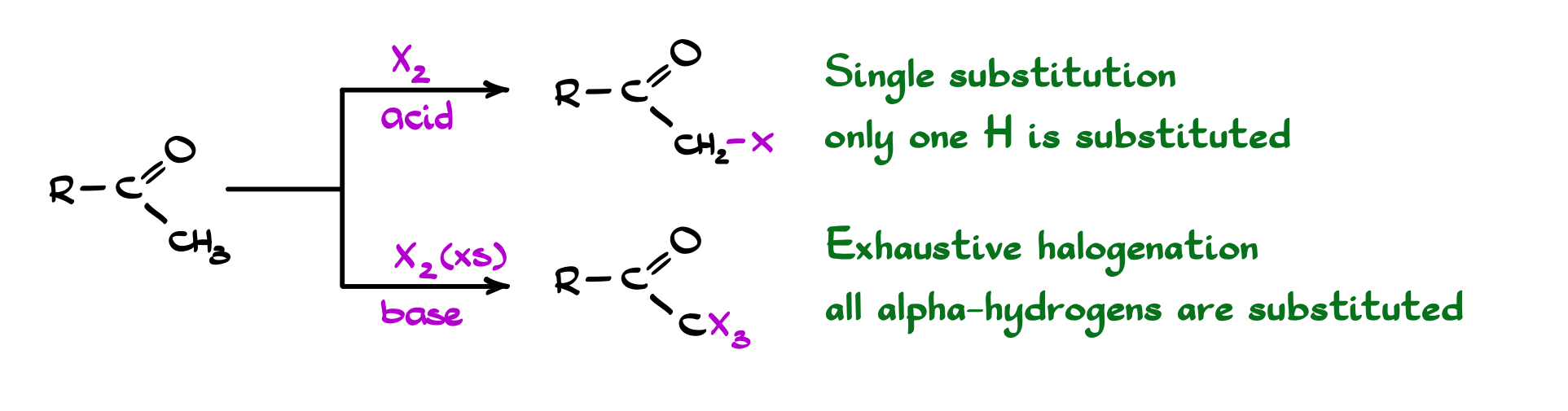
Halogenation of carbonyl compounds is one of the simplest ways to functionalize the α-position, but the mechanism and the outcome depend heavily on the conditions. Under acidic conditions, the reaction proceeds through the enol. The carbonyl first tautomerizes to the enol, and that electron-rich double bond then reacts with the halogen (Br₂, Cl₂, etc.). Because enol formation is reversible and relatively slow, this pathway is usually well-behaved and stops after monohalogenation. In other words, you get substitution of a single α-hydrogen, and the reaction is easy to control.
Under basic conditions, the story changes. Now we form the enolate, which reacts rapidly with the halogen. The key detail here is that each halogenation step makes the remaining α-hydrogens more acidic, which means the reaction doesn’t stop, it accelerates. As a result, you typically get polyhalogenation rather than a single substitution.
Haloform Reaction
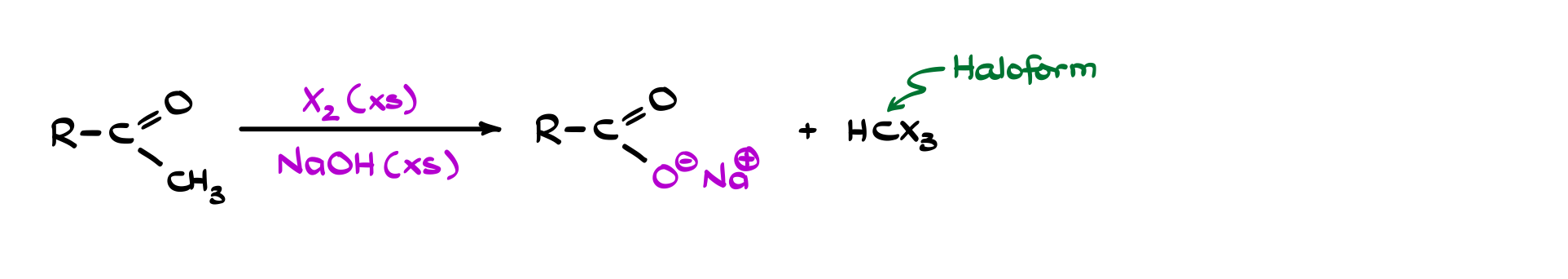
The haloform reaction is a classic extension of base-promoted α-halogenation, but with a very specific outcome. If you start with a methyl ketone (a carbonyl with a –COCH₃ group) and treat it with halogen (Cl₂, Br₂, or I₂) under basic conditions, the reaction doesn’t stop at simple substitution. Instead, all three α-hydrogens are replaced by halogens, giving a trihalomethyl ketone intermediate. At that point, the molecule essentially falls apart: hydroxide attacks the carbonyl, and the C–C bond next to it cleaves, producing a carboxylate and a haloform (CHX₃).
What makes this reaction particularly useful is how selective it is. It only works when that methyl group is present, so it’s often used as a diagnostic test for methyl ketones (and compounds that can be oxidized to them, like secondary alcohols with a –CH(OH)CH₃ fragment). Mechanistically, it’s a nice example of how repeated halogenation dramatically increases acidity and reactivity, pushing the system toward a cleavage that wouldn’t happen under milder conditions.
Alkylation of Carbonyls
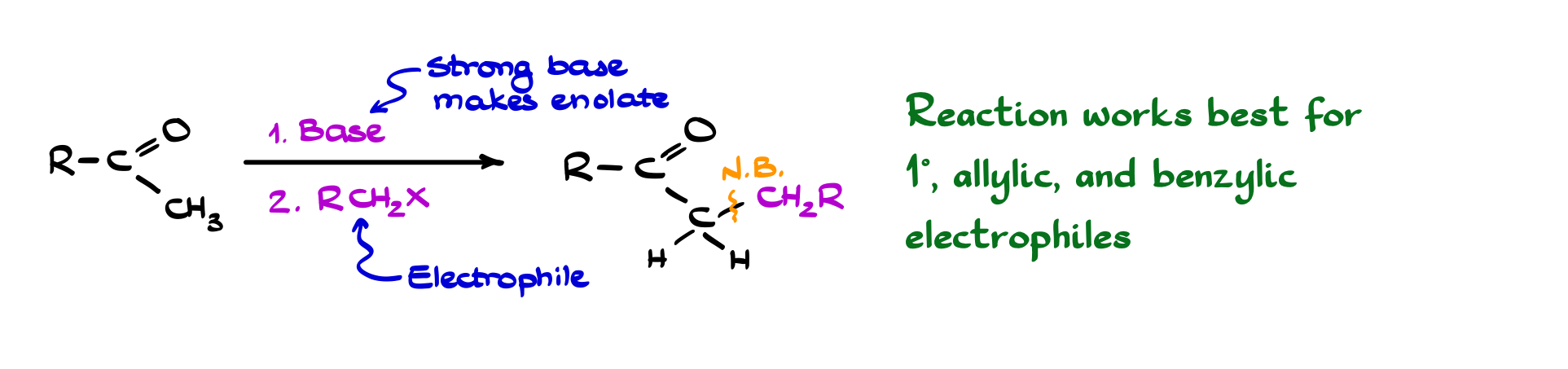
Once you form an enolate, one of the most straightforward things you can do is alkylate it. In practice, this means treating your ketone with a base to generate the enolate, and then reacting it with an alkyl halide. The enolate acts as a carbon nucleophile, attacking the electrophilic carbon of the alkyl halide in an SN2-type process and forming a new C–C bond at the α-position. Conceptually, it’s a simple and powerful idea: take a carbonyl compound, and extend its carbon skeleton by one piece at a time. Of course, the details matter. You generally need a primary alkyl halide to avoid elimination or competing side reactions, and the choice of base controls which enolate you form when there are multiple α-positions (kinetic vs thermodynamic control).
Aldol Addition and Aldol Condensation
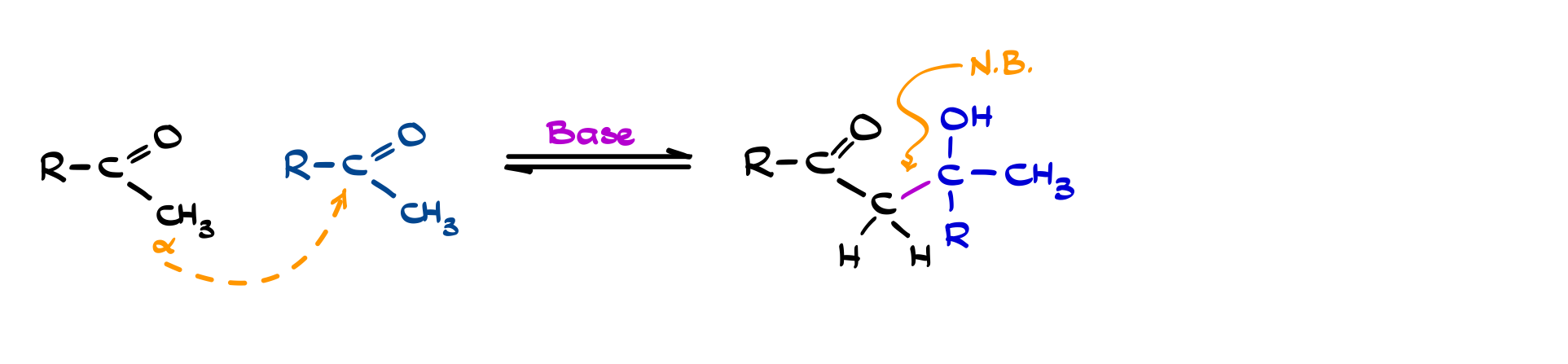
The aldol reaction is one of the central transformations built on enolate chemistry. When an enolate reacts with another carbonyl compound, it attacks the electrophilic carbonyl carbon and forms a new C–C bond, giving a β-hydroxy carbonyl: this is the aldol addition. Conceptually, it’s just nucleophilic addition, but instead of bringing in an external nucleophile, you’re using a carbonyl compound as both the nucleophile (via its enolate) and the electrophile. That’s what makes the reaction so powerful for stitching molecules together.
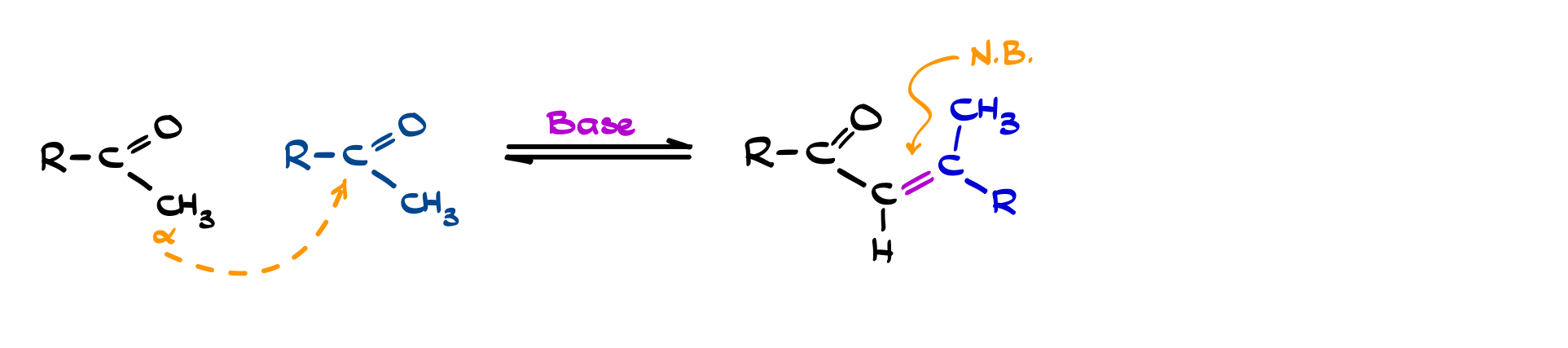
If you take it one step further, that β-hydroxy carbonyl can eliminate water to form an α,β-unsaturated carbonyl: this is the aldol condensation. Under basic or acidic conditions, dehydration is often favored, especially when the product gains conjugation, which stabilizes the system. So in practice, many aldol reactions don’t stop at the addition stage and go straight to the condensation product. Between these two steps, you’re not just forming a bond: you’re building extended π-systems in a predictable way, which is why aldol chemistry shows up everywhere from textbook problems to complex molecule synthesis.
Claisen and Dieckmann Condensations
The Claisen condensation is the ester analog of the aldol reaction, but with an important twist. Instead of forming a β-hydroxy carbonyl, you end up with a β-keto ester. Mechanistically, an enolate derived from an ester attacks another ester, forming a new C–C bond, followed by elimination of an alkoxide. Because esters are less reactive than aldehydes or ketones, you need a strong base that matches the leaving group (typically an alkoxide), and the reaction is driven forward by a final deprotonation of the product, which stabilizes the system and prevents reversal.
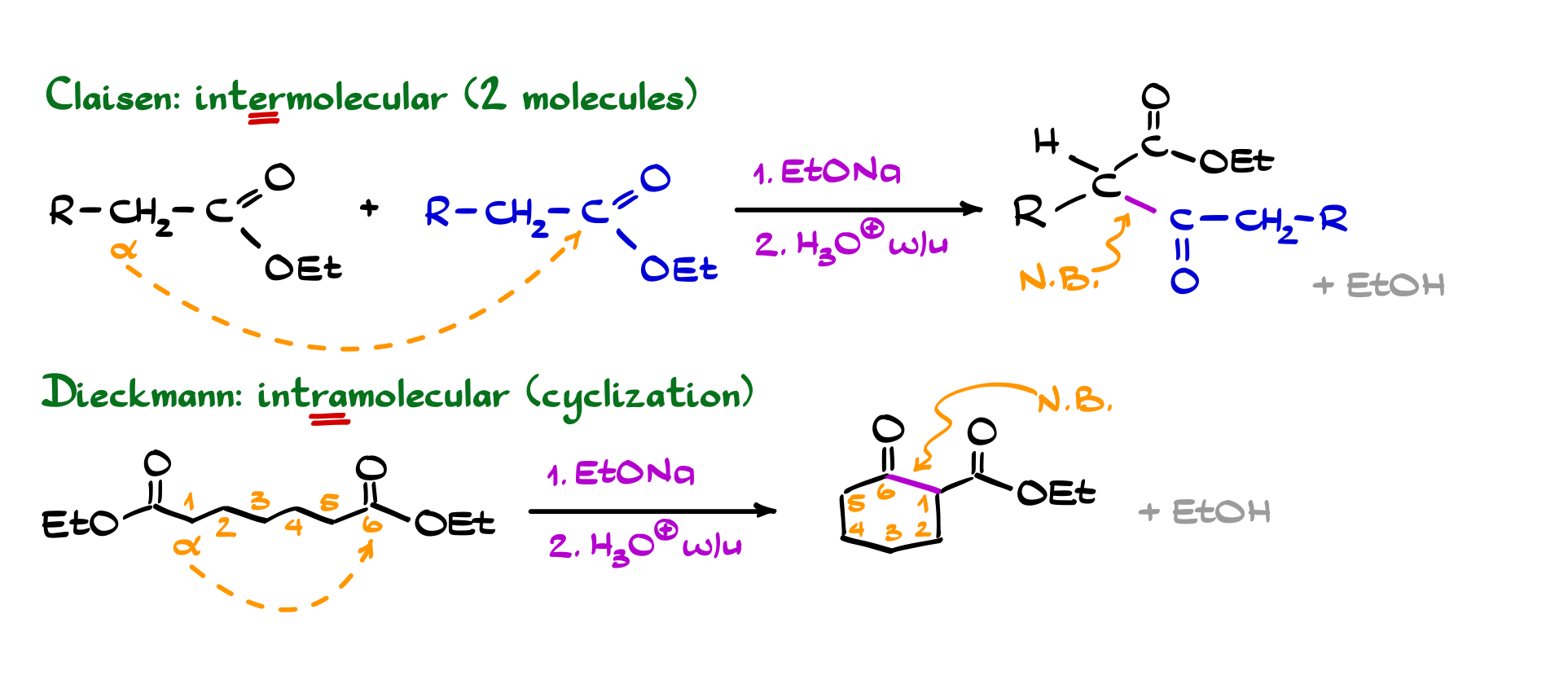
The Dieckmann condensation is simply the intramolecular version of the Claisen. When a molecule contains two ester groups positioned appropriately, the enolate can attack within the same molecule, forming a cyclic β-keto ester. This is a very efficient way to build rings, typically favoring 5- and 6-membered systems due to their stability. So while the Claisen reaction is about linking two separate pieces together, the Dieckmann is about folding a molecule onto itself to form a ring—same fundamental chemistry, just applied in a slightly different context.
Michael Addition
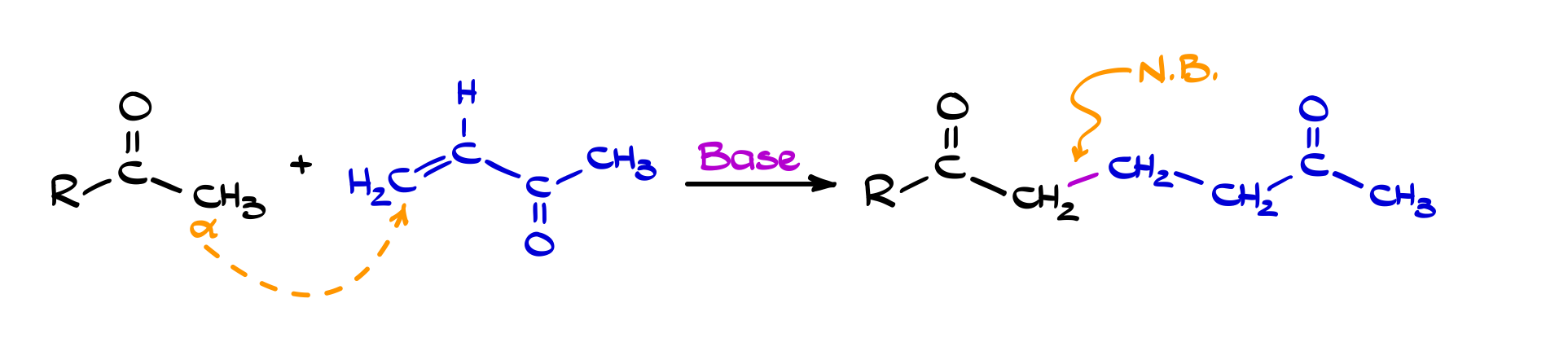
The Michael addition is what happens when an enolate stops behaving like a “hard” nucleophile and starts acting in a more controlled, conjugate (1,4-) addition sense. Instead of attacking directly at the carbonyl carbon, the enolate adds to the β-position of an α,β-unsaturated carbonyl compound. This gives you a new C–C bond while preserving the carbonyl, and after protonation, you end up with a 1,5-dicarbonyl-type system, which is an incredibly useful intermediate in synthesis.
What makes this reaction so valuable is its selectivity and compatibility. The conjugated system essentially “guides” the nucleophile to the β-carbon, especially under conditions that favor softer, more stabilized enolates. Compared to direct (1,2-) addition, the Michael addition is often more controlled and less aggressive, which is why it shows up so often in multi-step sequences. In practice, it’s one of the cleanest ways to extend a carbon framework while setting up further transformations down the line.
Robinson Annulation
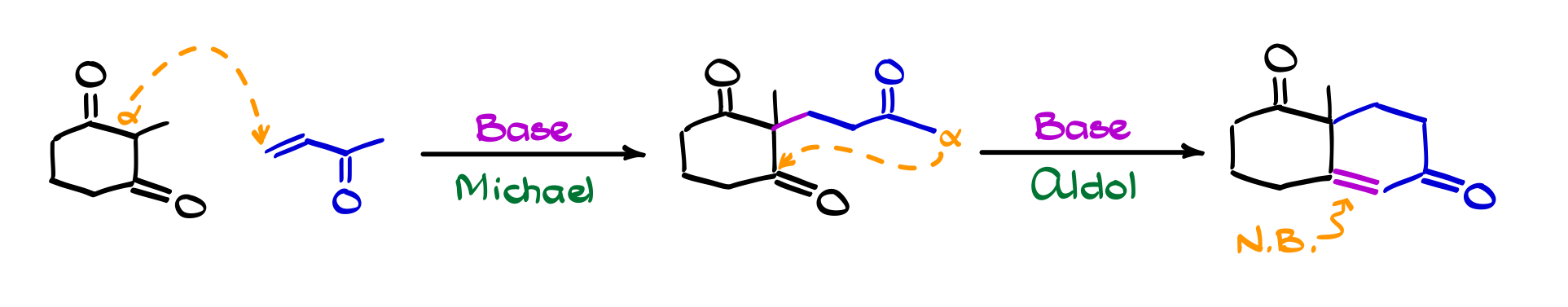
The Robinson annulation is a nice example of how a few familiar reactions can be chained together into something much more powerful. It combines a Michael addition with an intramolecular aldol condensation to build a six-membered ring with an α,β-unsaturated carbonyl system. In practice, an enolate first adds to an α,β-unsaturated carbonyl (the Michael step), giving a 1,5-dicarbonyl intermediate. That intermediate then forms a new enolate and cyclizes through an aldol reaction, followed by dehydration to give the final conjugated ring.
What makes the Robinson annulation so useful is how efficiently it constructs ring systems with built-in functionality. You’re not just closing a ring, you’re installing conjugation and setting up handles for further chemistry all in one sequence. That’s why this reaction shows up so often in synthesis, especially in the construction of complex cyclic molecules like steroids and terpenes. It’s a good reminder that once you understand the individual pieces: enolates, Michael additions, aldol reactions, you can start combining them in predictable ways to build much more sophisticated structures.
Now, when you have a basic idea of the chemistry of enols and enolates, go ahead and take a look at the detailed tutorials for each of the aspects of their chemistry below.